



Chorea-acanthocytosis (ChA) is an autosomal recessive disease caused by a mutation in the VPS13A gene, located on chromosome 9q21 and coding for the protein chorein. Chorein is a protein of 3000 amino acids, involved in the formation of the cell cytoarchitecture, exocytosis, and cell survival,1 especially in striatal neurons.2 Chorein deficit may activate autophagia and apoptotic mechanisms. It is expressed in erythrocytes, neurons, myocardiocytes, bone and muscle tissue, and peripheral nerves. Typical symptoms include psychiatric and cognitive alterations, tics, and the characteristic orofacial dystonia with biting and self-mutilation of the tongue, lips, and fingers. Patients may also present peripheral neuropathy, increased creatine-kinase (CK) levels, amyotrophy, and seizures.3
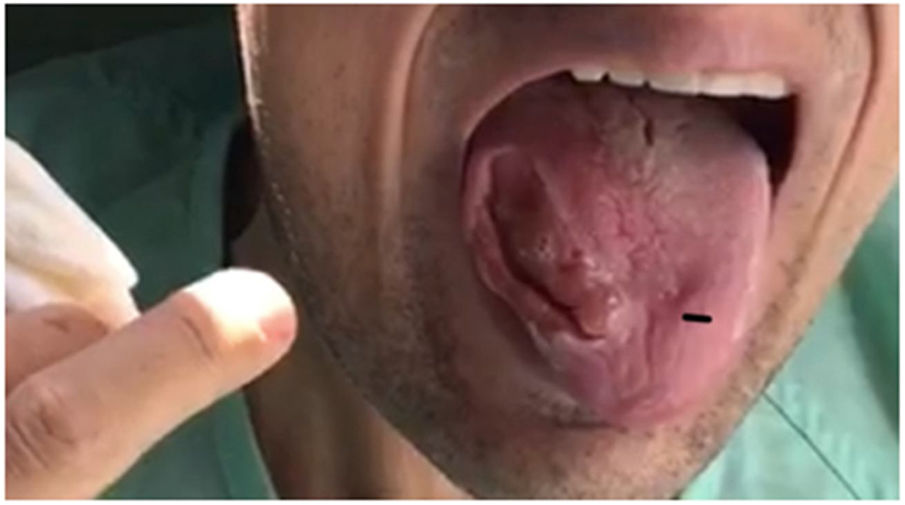
Our patient was a 28-year-old man, the second of 3 children born to consanguineous parents (first cousins) with no relevant personal history with the exception of adenoidectomy performed during childhood. His mother had been diagnosed with epilepsy and frontotemporal dementia. In 2011, he was under follow-up by the neurology and rheumatology departments due to high CK and adrenocorticotropic hormone levels detected in blood tests; the cause was not identified. In 2013, he presented 2 seizures, and treatment with levetiracetam was started; the patient decided to suspend treatment. In 2015, he began to bite his tongue when eating, presenting involuntary, dystonic orofacial movements since then with multiple episodes of tongue biting and a severe lesion to the right edge of the tongue (Fig. 1). Due to the inability to eat properly, he lost 40 kg over 2 years. In August 2017, he was admitted to the psychiatry ward due to obsessive-compulsive symptoms. During admission, the internal medicine department performed a study including laboratory tests for autoimmunity, toxic substances, serology tests, and a general blood test that revealed no alterations except for elevated CK levels. An electroencephalography and a magnetic resonance imaging study returned normal results. Due to a new episode of tongue biting, the patient developed symptoms of sepsis, resulting in admission to the intensive care unit. Once he stabilised, he was transferred to the neurology department for further study. A motor nerve conduction study revealed sensorimotor axonal polyneuropathy and a peripheral blood smear test showed echinocytes. During admission he presented another seizure, with oxygen desaturation due to aspiration of the gauzes he used to avoid the constant tongue biting; therefore, treatment with eslicarbazepine acetate was started. To control behaviour and dystonic oral movements, we tried several drugs (clonazepam, haloperidol, tetrabenazine, pimozide, paliperidone, olenzapine, aripiprazole, naltrexone, etc), with little response and variable tolerance. Botulinum toxin infiltrations were administered to the temporal, pterygoid, masseter, and genioglossus muscles to control tongue biting.

Given the compatible symptoms and the clearly degenerative progression, we requested a genetic panel for ChA, which revealed homozygous presence of a mutation of the VPS13A gene (c.2347C>T), causing substitution of glutamine with a premature stop codon at position 783. This mutation has not previously been described, although it is pathological in our patient.
ChA is a rare disorder of autosomal recessive inheritance,4 whose initial symptoms may be mistaken for purely psychiatric symptoms. It should be considered in patients with compatible symptoms, especially if there is family history of consanguineous sexual relationships. Definitive diagnosis is genetic, since presence of acanthocytosis in peripheral blood is not constant.5
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Garrido-Fernández A, Santos-Lasaosa S, Bellosta-Diago E, Sáchez-Valiente S. Nueva mutación patogénica de corea-acantocitosis. Neurología. 2020;35:73–74.
