




Neuromyelitis optica spectrum disorders (NMOSD) are a group of autoimmune inflammatory diseases of the central nervous system, most frequently affecting the optic nerve and spinal cord. The discovery of aquaporin-4 antibodies (AQP4-ab) has contributed to the understanding of manifestations not affecting these structures, and new diagnostic criteria have been developed to facilitate early, accurate diagnosis, both for seropositive and seronegative phenotypes.1
Presentation of NMOSD as a tumefactive demyelinating lesion (TDL) in isolation, without spinal cord or optic nerve involvement, is extremely rare.
We report a case of NMOSD presenting with TDL only, and review similar cases from the literature.
PatientThe patient was a 62-year-old woman with no relevant history who consulted due to a 2-week history of dysarthria, aphasia, and right hemiparesis. A brain MRI study (Fig. 1, series 1) showed an extensive frontotemporal gadolinium-enhancing lesion, suggestive of TDL or an atypical high-grade glial tumour. The results of an 11C-methionine PET/CT study were not indicative of neoplastic origin. Cerebrospinal fluid analysis detected mild pleocytosis (8 cells/mL), normal protein levels (0.51 g/L), absence of oligoclonal bands, and normal cytology and immunophenotyping findings. Spinal MRI and visual evoked potentials yielded normal results. Testing for antinuclear antibodies returned positive results (1/1280), with a pattern of antimitochondrial antibodies, compatible with primary biliary cholangitis. We administered 2 megaboluses of intravenous methylprednisolone (1 g), followed by oral prednisolone and rehabilitation therapy.
 Brain MRI: axial T2-weighted and coronal FLAIR sequences (1a and b) showing a large left frontal periventricular lesion, extending to the lenticular nucleus and insula, with a minimal mass effect on the lateral ventricle. The gadolinium-enhanced T1-weighted sequence (1c) shows irregular peripheral “cloud-like” enhancement.2 The diffusion sequence (1d and e) reveals mild restriction in the medial area, which is not suggestive of an ischaemic lesion. Series 2) Brain MRI: axial and coronal T2-weighted and FLAIR sequences showing a reduction in the size of the previous lesion (2a), and 2 new lesions: one extending towards the right basal ganglia (2b) and another predominantly left-sided lesion surrounding the third ventricle and involving the hypothalamus (2c); both lesions are gadolinium-enhancing (2d and e). Series 3) Left: spinal cord MRI. Sagittal gadolinium-enhanced T1-weighted and T2-weighted sequences (3a–c) showing multiple gadolinium-enhancing lesions, the largest extending from C4 to T2. Axial T2-weighted and gadolinium-enhanced T1-weighted sequences (3d and e) reveal heterogeneous lesions with areas of hyperintensity and hypointensity. Right: brain MRI. axial FLAIR and gadolinium-enhanced T1-weighted sequences (3f and g) show multiple new hyperintensities involving the periventricular area, corpus callosum, and left occipital lobe; most lesions present gadolinium enhancement.")
Series 1) Brain MRI: axial T2-weighted and coronal FLAIR sequences (1a and b) showing a large left frontal periventricular lesion, extending to the lenticular nucleus and insula, with a minimal mass effect on the lateral ventricle. The gadolinium-enhanced T1-weighted sequence (1c) shows irregular peripheral “cloud-like” enhancement.2 The diffusion sequence (1d and e) reveals mild restriction in the medial area, which is not suggestive of an ischaemic lesion. Series 2) Brain MRI: axial and coronal T2-weighted and FLAIR sequences showing a reduction in the size of the previous lesion (2a), and 2 new lesions: one extending towards the right basal ganglia (2b) and another predominantly left-sided lesion surrounding the third ventricle and involving the hypothalamus (2c); both lesions are gadolinium-enhancing (2d and e). Series 3) Left: spinal cord MRI. Sagittal gadolinium-enhanced T1-weighted and T2-weighted sequences (3a–c) showing multiple gadolinium-enhancing lesions, the largest extending from C4 to T2. Axial T2-weighted and gadolinium-enhanced T1-weighted sequences (3d and e) reveal heterogeneous lesions with areas of hyperintensity and hypointensity. Right: brain MRI. axial FLAIR and gadolinium-enhanced T1-weighted sequences (3f and g) show multiple new hyperintensities involving the periventricular area, corpus callosum, and left occipital lobe; most lesions present gadolinium enhancement.
Seven months later, after a respiratory infection, the patient presented symptoms of confusion, worsening of the right hemiparesis, and syndrome of inappropriate antidiuretic hormone secretion (SIADH). A brain MRI study (Fig. 1, series 2) revealed additional gadolinium-enhancing lesions, showing a cloud-like enhancement pattern. Indirect immunofluorescence testing for AQP4-IgG-ab returned positive findings. The patient was treated with intravenous methylprednisolone, plasmapheresis, and cyclophosphamide.
Three months later, and after 3 cycles of cyclophosphamide, she presented spinal cord syndrome. An MRI study (Fig. 1, series 3) showed longitudinally extensive myelitis and multiple gadolinium-enhancing lesions. Intravenous methylprednisolone was administered once more, and we added rituximab based on the regular assessment of memory B cells.3
The patient remains clinically stable at 36 months of follow-up, after 5 cycles of rituximab; radiological signs have improved considerably.
DiscussionThe radiological characteristics of the baseline brain lesion led us to perform differential diagnosis between TDL and atypical glial tumour4; however, we subsequently came to suspect inflammatory aetiology due to the detection of antibodies associated with another autoimmune disease (despite the absence of symptoms) and the negative findings of the 11C-methionine PET/CT study to determine possible neoplastic origin. The favourable clinical and radiological response to corticotherapy further supported this diagnostic hypothesis, and we opted not to perform a brain biopsy. Following the second episode, consisting of diencephalic involvement (SIADH), the detection of AQP4-ab, and the exclusion of alternative diagnoses, we diagnosed the patient with NMOSD. This form of onset is extremely rare, although the longitudinally extensive myelitis that the patient ultimately developed is typical of the disease.
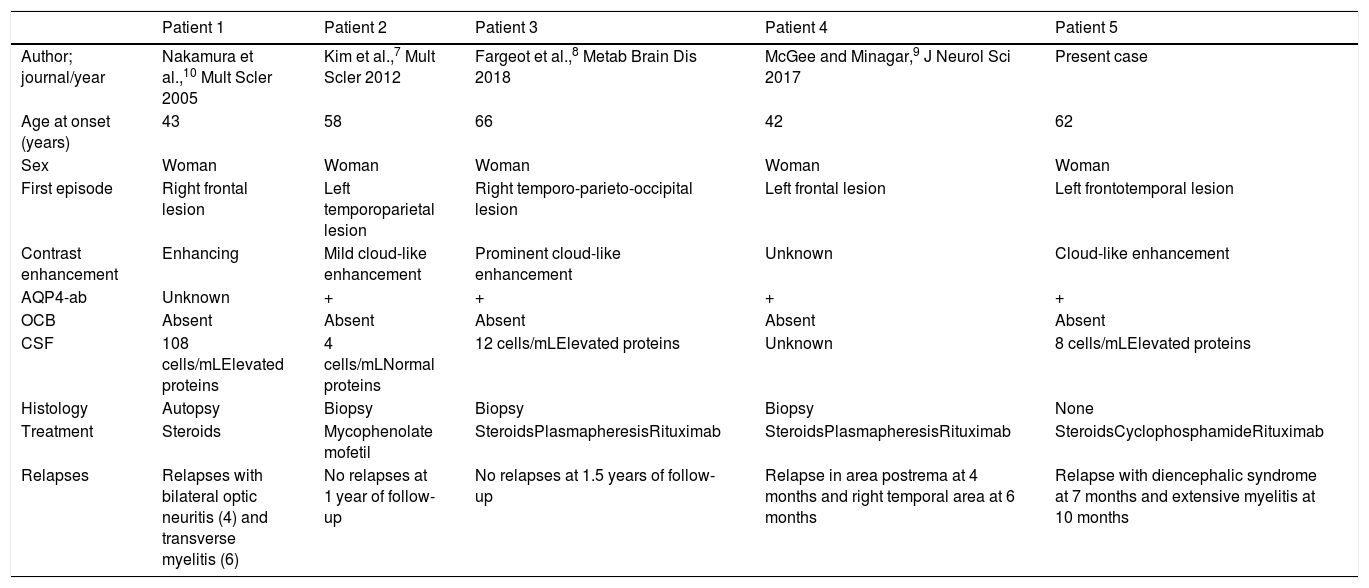
Only 15% of patients with NMOSD present brain involvement at onset, with or without associated neurological deficits, and onset with TDL in isolation is extremely rare.5,6 A literature search identified 4 cases of NMOSD presenting with TDL only.5,7–10 All 4 patients were women, with a similar age of onset to that of our patient and variable initial symptoms; radiology studies revealed cloud-like enhancement in 2 cases.2 All 4 patients showed absence of oligoclonal bands, and 3 were positive for AQP4-ab. Two presented multiple relapses during the first month of follow-up. Two patients remained stable after treatment with rituximab, as was the case with our patient (Table 1).
Clinical cases of neuromyelitis optica spectrum disorder presenting with tumefactive demyelinating brain lesions in isolation.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Author; journal/year | Nakamura et al.,10 Mult Scler 2005 | Kim et al.,7 Mult Scler 2012 | Fargeot et al.,8 Metab Brain Dis 2018 | McGee and Minagar,9 J Neurol Sci 2017 | Present case |
| Age at onset (years) | 43 | 58 | 66 | 42 | 62 |
| Sex | Woman | Woman | Woman | Woman | Woman |
| First episode | Right frontal lesion | Left temporoparietal lesion | Right temporo-parieto-occipital lesion | Left frontal lesion | Left frontotemporal lesion |
| Contrast enhancement | Enhancing | Mild cloud-like enhancement | Prominent cloud-like enhancement | Unknown | Cloud-like enhancement |
| AQP4-ab | Unknown | + | + | + | + |
| OCB | Absent | Absent | Absent | Absent | Absent |
| CSF | 108 cells/mLElevated proteins | 4 cells/mLNormal proteins | 12 cells/mLElevated proteins | Unknown | 8 cells/mLElevated proteins |
| Histology | Autopsy | Biopsy | Biopsy | Biopsy | None |
| Treatment | Steroids | Mycophenolate mofetil | SteroidsPlasmapheresisRituximab | SteroidsPlasmapheresisRituximab | SteroidsCyclophosphamideRituximab |
| Relapses | Relapses with bilateral optic neuritis (4) and transverse myelitis (6) | No relapses at 1 year of follow-up | No relapses at 1.5 years of follow-up | Relapse in area postrema at 4 months and right temporal area at 6 months | Relapse with diencephalic syndrome at 7 months and extensive myelitis at 10 months |
AQP4-ab: aquaporin 4 antibodies; CSF: cerebrospinal fluid; NMOSD: neuromyelitis optica spectrum disorders; OCB: oligoclonal bands; TDL: tumefactive demyelinating lesion.
According to the published series, MRI detects brain lesions at onset in 43%-70% of patients, increasing to 80% as the disease progresses, although the majority of lesions are usually non-specific.5,6 Kim et al.5 establish 5 specific categories of brain MRI findings in NMOSD, including extensive hemispheric lesions, a rare form of presentation. At onset, they appear in the context of an acute disseminated encephalomyelitis–like disorder, myelitis, and/or optic neuritis; it is extremely unusual for them to appear in isolation.
TDLs are a diagnostic challenge and usually require histological analysis. A proper diagnostic approach may avoid the need for unnecessary invasive procedures, such as biopsy. Studies into the radiological characteristics of TDLs and the reporting of cases of this rare form of onset of NMOSD will assist us in diagnosing our patients. To our knowledge, this is the first case diagnosed without a histology study.
Please cite this article as: Zhu N, Presas-Rodríguez S, Núñez-Marín F, Quirant-Sánchez B, Ramo-Tello C. Lesión pseudotumoral única, una presentación de inicio infrecuente del trastorno del espectro de la neuromielitis óptica. Neurología. 2021;36:396–398.
