



Tapia syndrome is defined as paralysis of one side of the tongue and the ipsilateral vocal cord, with preserved soft palate motility, secondary to concurrent lesions to the hypoglossal and vagus nerves (the twelfth and tenth cranial nerves, respectively).
Antonio García Tapia, a Spanish otorhinolaryngologist, first described the syndrome in 1904 in a patient with an upper neck injury from a bull’s horn (“matador’s disease”).1 However, the eponym has been used in reference to a crossed syndrome of dorsal medullary origin (with involvement of the hypoglossal and ambiguus nuclei and the pyramidal tract),2,3 and to describe peripheral involvement of the vagus and hypoglossal nerves at the cervical level.3
The peripheral form has been observed after manipulation of the airway for orotracheal intubation.
We present the case of a 70-year-old man with history of dilated cardiomyopathy secondary to alcohol abuse, who was admitted to our hospital due to cardiorespiratory arrest.
The cause of the attack was ventricular fibrillation; the patient required emergency life support for 15 minutes, with basic and advanced cardiopulmonary resuscitation. The attack occurred in a public space, and emergency intubation was performed before the patient was transferred to our hospital. Upon arrival at the emergency department, the patient was admitted to the coronary care unit, where an emergency coronography was performed and a hypothermia protocol was initiated (target temperature, 33 °C). The patient was kept under sedation during the first days after arrival at hospital, with no initial neurological assessment being performed at this time. Incidentally, a second orotracheal intubation procedure was required at 72 hours following obstruction of the first intubation tube.
As the patient continued to need mechanical ventilation a week after admission, a tracheostomy was performed to prevent airway lesions associated with orotracheal intubation. At this point, the patient continued to require sedation and parenteral feeding. Ten days after admission to the coronary care unit, the patient began to recover consciousness, spontaneously opening his eyes; brainstem reflexes were preserved, including the pupillary reflex, oculocephalic reflexes, and the cough reflex. Mechanical ventilation was suspended at 2 weeks and a silver tracheostomy tube was placed.
Two weeks after the attack, an MRI study was performed as part of the coronary care unit’s protocol for the assessment of brain damage in patients recovering from cardiorespiratory arrest. The study identified no acute or chronic ischaemic lesions in any region of the brain; electroencephalography and somatosensory evoked potentials studies yielded no relevant findings.
The patient remained in the coronary care unit for the following 2 weeks, showing satisfactory progression: level of consciousness remained adequate at all times and he gradually recovered limb strength and mobility, deglutition, and phonation. Dysphagia and dysphonia were not observed in early assessments. The patient’s progression was considered to be excellent, and he was transferred to the cardiology inpatient ward one month after the cardiorespiratory arrest. The initial assessment conducted by the cardiology department included a basic neurological examination, which detected reduced tongue mobility and hypophonia, with no other focal neurological signs and a normal level of consciousness.
In the light of these findings, the patient underwent a comprehensive assessment by the neurology department, which revealed dysphonia and hypophonia and paralysis of the left half of the tongue without atrophy. We identified no other findings suggesting involvement of the brainstem, pyramidal tracts, or long sensory pathways.
The otorhinolaryngology department performed further testing of the patient, and a fibre-optic laryngoscopy study identified vocal cord paralysis in adduction on the left side. A second MRI study ruled out central origin of the symptoms, as well as space-occupying lesions in the skull base.
The patient was diagnosed with peripheral Tapia syndrome secondary to trauma occurring after repeated orotracheal intubation; we opted for conservative treatment with speech and swallowing rehabilitation. Symptoms improved gradually, fully resolving 4 months after onset.
This is an illustrative case of Tapia syndrome of probable peripheral origin: in this patient, we suspect traumatic neuropathy involving the tenth and twelfth cranial nerves, probably due to repeated orotracheal intubation.
It is important to be familiar with the anatomy of the most common syndromes affecting the brainstem and the emergence of the cranial nerves, both in the skull base and on their course along the upper cervical spine. In the case of Tapia syndrome, both cranial nerves (X and XII) emerge from the medulla oblongata, passing through the skull base to the lateral pharyngeal or poststyloid space.
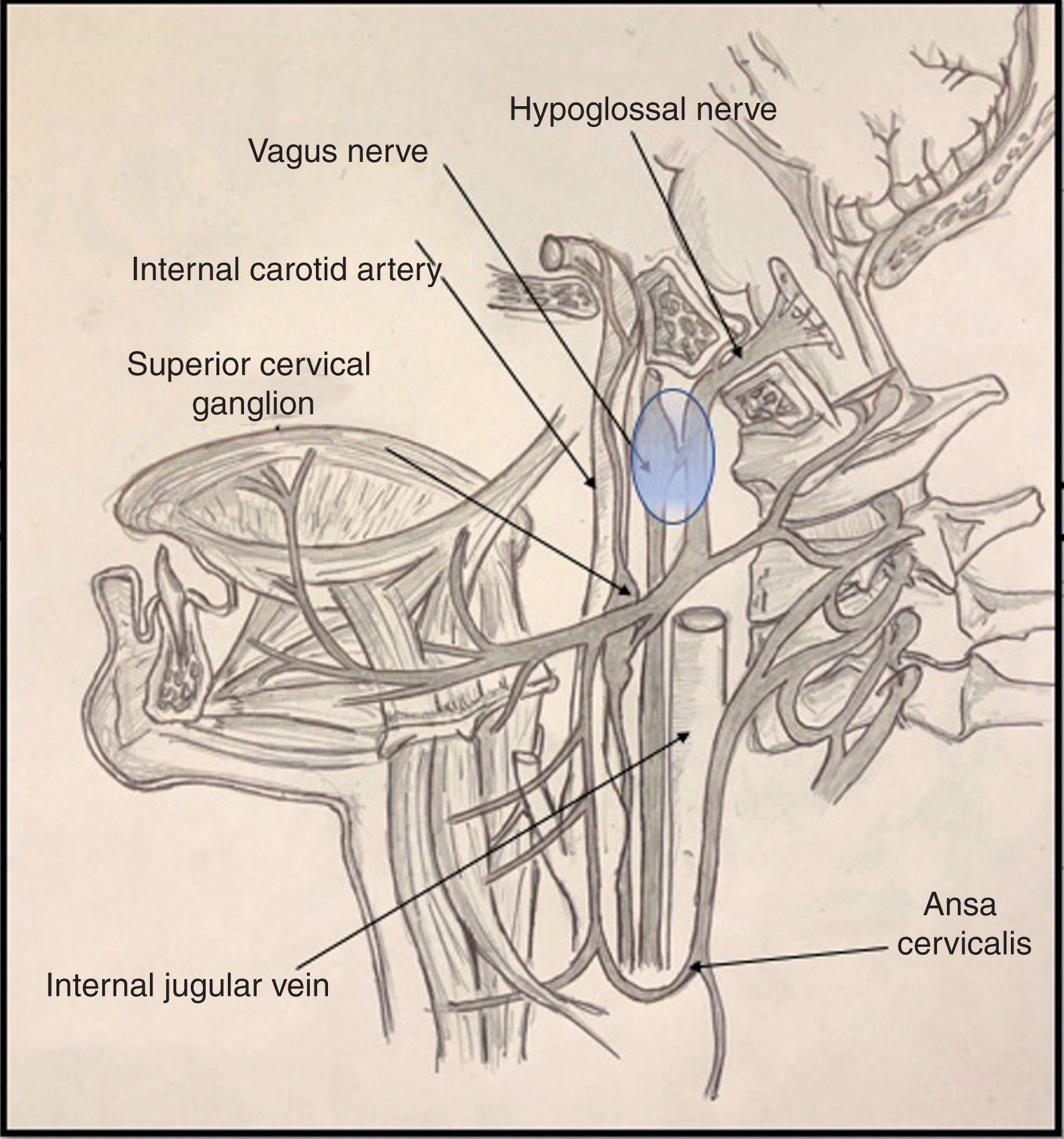
Cranial nerve X passes through the anterior compartment of the jugular foramen, alongside the glossopharyngeal and accessory nerves (nerves IX and XI) and the jugular vein, to the poststyloid space. Upon entering the poststyloid space, the nerve joins the carotid sheath, where it runs posterior to the internal carotid artery and the internal jugular vein. At this level, the most important structure of the nerve is the inferior (or nodose) ganglion, whose inferior part gives rise to the superior laryngeal nerve.
Cranial nerve XII presents a short trajectory through the subarachnoid space, then passes through the hypoglossal canal into the poststyloid space. There, it initially descends posterior to the internal carotid artery. It reaches 2 structures of anatomical interest: the upper part of the superior cervical ganglion of the sympathetic trunk, and then the posterior segment of the nodose ganglion, to which it adheres (Fig. 1). Subsequently, the nerve changes direction, travelling ventrally then ramifying in its final (suprahyoid) segment.4

Various proposed mechanisms of nerve trauma may explain the combined involvement of the tenth and twelfth cranial nerves during orotracheal intubation. The first mechanism is related to the crossing of the hypoglossal and vagus nerves at the nodose ganglion (Fig. 1). Here, these structures are in close proximity to the transverse process of the C1 vertebra; the sustained hyperextension of the neck needed for proper intubation may explain the direct trauma to both nerves, due to contact against these bone structures.5,6
The other mechanism proposed is a direct lesion simultaneously involving the suprahyoid portion of the hypoglossal nerve and the recurrent laryngeal nerve at the level of the hypopharynx (pyriform sinus). According to this hypothesis, the passage of the intubation tube through the oropharynx would compress the hypoglossal nerve against the greater cornu of the hyoid, and the subsequent inflation of the pilot balloon would cause the recurrent laryngeal nerve to contact the posteromedial edge of the thyroid cartilage, constituting a mechanism of direct trauma.7
Treatment for Tapia syndrome secondary to orotracheal intubation is fundamentally supportive, with speech and swallowing rehabilitation; the use of intravenous corticotherapy with dexamethasone in the first 10 to 14 days is also accepted. One article even proposes a classification of the syndrome according to the grade of difficulty swallowing.8
Both the patient’s improvement with conservative treatment and the complete resolution of his symptoms indicate neurapraxia secondary to axonal damage as the probable aetiology; neurotmesis is less probable, as the damage to both nerves would have been irreversible. Furthermore, neurapraxia is the proposed aetiological mechanism in most of the reported cases of Tapia syndrome secondary to orotracheal intubation.9
Please cite this article as: Silva-Hernández L, Gil Rojo C, González García N, Porta-Etessam J. Síndrome de Tapia tras intubación orotraqueal: a propósito de un caso. Neurología. 2020;35:421–423.
