



La titina (TTN) es una proteína sarcomérica que se puede encontrar en los músculos cardíaco y esquelético. Está codificada por un único gen, ubicado en el brazo largo del cromosoma 21. Las mutaciones del gen TTN se han asociado con diversas miocardiopatías y trastornos del músculo esquelético2. Un amplio espectro de enfermedades del músculo esquelético se han relacionado con mutaciones de este gen (distrofia muscular tibial de inicio tardío, titinopatía distal recesiva de inicio temprano, distrofia muscular de cinturas autosómica recesiva tipo 2J, entre otros)2. Entre ellas destaca la distrofia del músculo tibial (DMT), también conocida como miopatía distal de Udd (inicio tardío, tipo IIa). Se caracteriza por un inicio tardío (entre 35-55 años), evolución lenta y generalmente limitada a los músculos del compartimento anterior distal de las extremidades inferiores3. Los valores de creatinina quinasa sérica son normales o ligeramente elevados y los datos del electromiograma (EMG) son de miopatía3,4. En la biopsia muscular los hallazgos incluyen variabilidad del tamaño de las fibras, núcleos centrales, necrosis, presencia de tejido fibroadiposo y vacuolas con borde5. La resonancia magnética (RM) muscular pone de manifiesto el reemplazo graso de manera selectiva en los músculos de los compartimentos anteriores de los miembros inferiores.
Presentamos el caso de un varón de 37 años con clínica de debilidad de miembros inferiores (MMII) y atrofia muscular dependiente de la musculatura peroneal, tibial y extensora de los dedos del pie. Entre sus antecedentes personales no había historial de diabetes mellitus, enfermedades oncológicas o trastornos del tejido conectivo. Simultáneamente se detecta comorbilidad cardíaca y es diagnosticado de miocardiopatía dilatada (MCD) de causa no isquémica. En la historia familiar disponible, no hay datos de consanguinidad y destaca la presencia de enfermedad muscular no filiada en un tío materno con debilidad y atrofia muscular en MMII. Hasta la actualidad, los progenitores no han consultado por sintomatología neurológica o cardíaca.
El examen neurológico mostró atrofia y debilidad del compartimento anterior de ambos MMII, con disminución de fuerza para la dorsiflexión de forma asimétrica, derecho (1/5 en la escala Medical Research Council) e izquierdo (3/5). En la evaluación de la marcha, imposibilidad para deambular de talones por debilidad en la dorsiflexión de ambos pies.
La analítica de sangre incluyendo enzimas musculares y estudio de cadena respiratoria mitocondrial fueron normales. El estudio electroneurográfico fue normal y en el electromiograma se registraron potenciales de unidad motora polifásicos de pequeña amplitud con reclutamiento precoz de los músculos tibiales anteriores, compatible con un proceso miopático crónico. La biopsia del músculo cuádriceps reveló alteraciones miopáticas con centralización nuclear y degeneración vacuolar con mínimo infiltrado inflamatorio endomisial, sin presencia de fibras rojas rasgadas y con tinción COX normal.
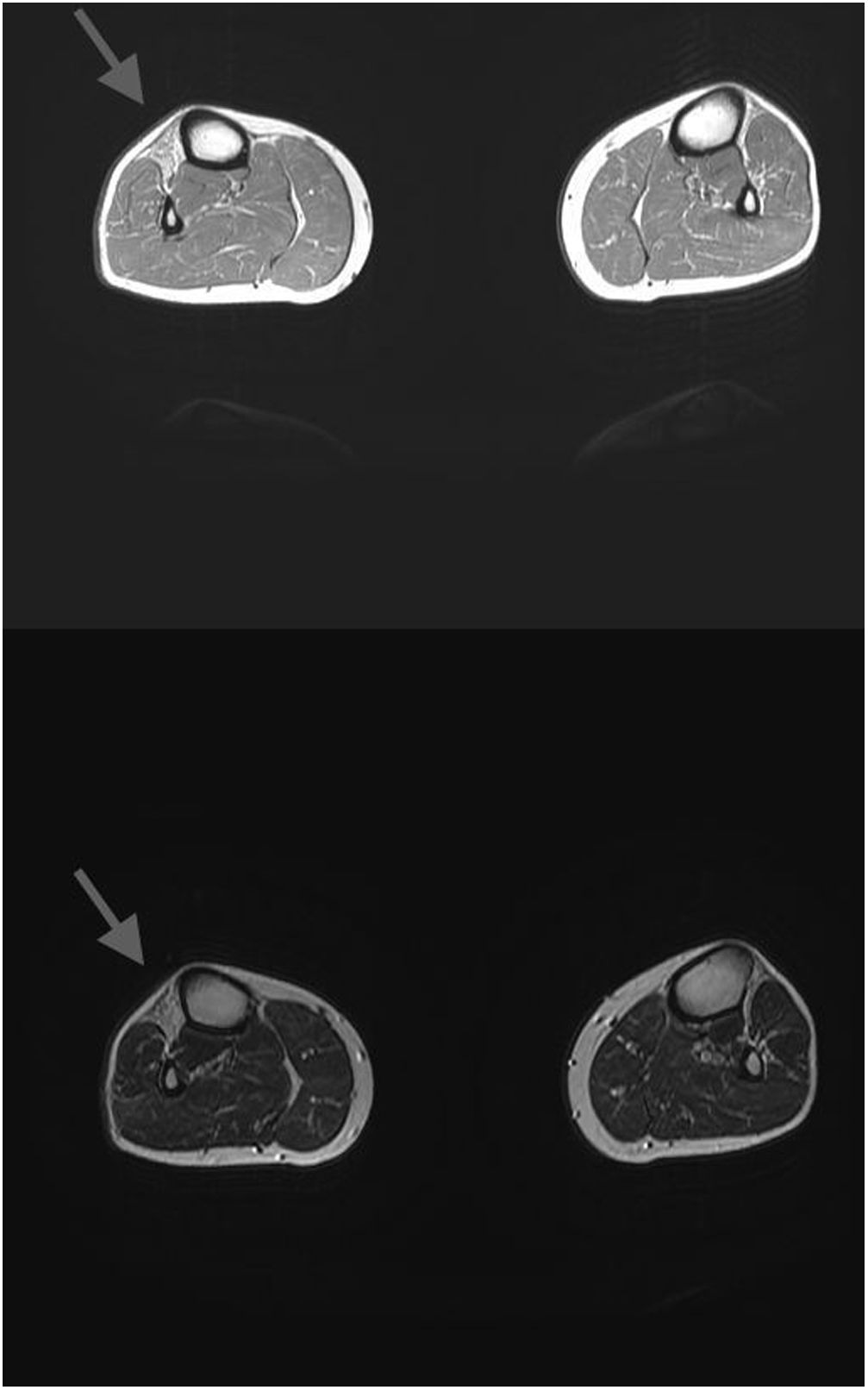
La RM muscular de los MMII mostraba atrofia con sustitución grasa de la musculatura extensora (tibialis anterior, extensor digitorum longus y extensor hallucis longus de predominio derecho) (fig. 1). Asimismo, se completó el estudio con una RM cardíaca donde se apreciaba dilatación bi-ventricular con disfunción sistólica moderada de ambos ventrículos y dilatación de ambas aurículas compatible con MCD de etiología no isquémica.

Mediante secuenciación masiva se realizó un estudio de 8 genes asociados a distrofias musculares (ANO5, DMD, DYSF, EMD, FHL1, LMNA, SYNE1 y SYNE2) sin encontrar ninguna mutación patogénica. Posteriormente, se completó el estudio mediante secuenciación completa de 22 genes, incluido el gen TTN, lo que reveló una pérdida de heterocigosidad que podría afectar al menos a los exones 32 a 364 del gen. Se identificó una variante homocigota c.86581T>A (p.Trp28861Arg) en el gen TTN. En el estudio de cosegregación, esta variante fue detectada en heterocigosidad en ambos progenitores.
Las mutaciones en el gen TTN se han asociado con diferentes enfermedades musculares, cardiomiopatías o combinaciones de estas. La primera mutación (inserción-eliminación de 11 pb) se informó en 2002 y se denominó FINmaj6. Posteriormente, se identificó una variante dominante de sentido erróneo en el exón 364 (c.107840T>A p.Ile35947Asn) en una familia belga5. En 2008 Hackman et al., descubrieron 3 nuevas mutaciones en 2 familias españolas y 2 familias francesas (2 deleciones, g.292998delT y g.293376delA) y una mutación sin sentido g.293379C>T (p.Q33396X)4. Posteriormente, se describió el fenotipo de una familia italiana con DMT y una nueva mutación heterocigota 293326A>C que predice un cambio His33378Pro7. Algahtani et al. describieron una mutación sin sentido heterocigótica c.85652C>G, p. (Pro28551Arg) en un paciente saudí con debilidad facial bilateral8.
Presentamos el caso clínico de una nueva variante en homocigosis en el gen TTN en un paciente español con DMT y MCD. La muestra del paciente presenta la variante nucleotídica c.86581T>A (p.Trp28861Arg) en aparente homocigosis en el exón 341 del gen TTN. Esta variante implica un cambio de triptófano a arginina en la posición 28861 de la cadena polipeptídica, aminoácidos con diferencias fisicoquímicas moderadas. El cambio se encuentra en un residuo que forma parte del dominio fibronectina tipo III, ampliamente conservado en proteínas animales. Esta variante no ha sido descrita previamente en ninguna publicación ni aparece en las bases de datos ClinVar, dbSNP, ExAC o ESP.
Esta nueva variante en homocigosis en el gen TTN, no descrita en la literatura revisada, aunado a un fenotipo clínico de debilidad de compartimento anterior en miembros inferiores sugiere el origen patogénico de la mutación, que puede estar relacionada con genes causantes de DMT y MCD.

