



Spinocerebellar ataxia autosomal recessive (SCAR) represents a heterogeneous chronic and progressive neurological diseases group. They usually occur at an early age in a progressive manner. Diagnosis is complex due to phenotypic overlap. SCARs account for more than 50% of all ataxia cases of genetic origin, with a prevalence of 3–4/100 000. According to international published series, Friedreich's ataxia (FA) is the most common. In Mexico, more than 90% of patients with suspected SCAR remain without etiologic diagnosis after ruling out FA and acquired causes of ataxia. Our main goal was to reach a diagnosis using genomic tools in this group of patients.
Materials and methodsAt the National Institute of Genomic Medicine, we used next-generation sequencing as a diagnostic tool in 4 patients with a clinical diagnosis of SCAR to identify and classify etiologic variants responsible for this group of disorders.
ResultsTwo novel pathogenic variants were identified in the SACS gene, and the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) was established.
ConclusionsThis is the first report of spastic ataxia of Charlevoix-Saguenay cases in Mexico.
Las ataxias espinocerebelosas autosómico recesivas (SCAR) representan un grupo heterogéneo de enfermedades crónicas progresivas. Este grupo de enfermedades inician a una edad temprana y suelen ser progresivas. Su diagnóstico es complejo debido a superposición fenotípica. Las SCAR representan más del 50% de los casos de ataxias de origen genético con una prevalencia de 3–4/100,000, siendo la ataxia de Friedreich (FA) la más común de acuerdo con series internacionales publicadas. En México, después de descartar FA, más del 90% de pacientes con sospecha de SCAR quedsn sin diagnóstico etiológico. Nuestro principal objetivo es el de establecer un diagnóstico etiológico mediante el uso de herramientas genómicas en este grupo de pacientes.
Material y métodosEn el Instituto Nacional de Medicina Genómica, utilizamos secuenciación de siguiente generación como herramienta diagnóstica en 4 pacientes con SCAR para identificar y clasificar las variantes patogénicas responsables del cuadro clínico.
ResultadosIdentificamos dos variantes nuevas en el gen SACS y establecimos el diagnóstico de ataxia espástica recesiva de Charlevoix-Saguenay (ARSACS).
ConclusionesEste es el primer reporte de ataxia espástica recesiva de Charlevoix-Saguenay en México.
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) was first reported by JP Bouchard in 19781 among the population from the Charlevoix-Saguenay-Lac-Saint-Jean (CSLSJ) region of Quebec, Canada, where the estimated carrier prevalence of the original mutation (c.8844delT) is 1/22 due to a founder effect in the French-Canadian population.2
ARSACS is considered a rare recessive disease occurring worldwide.3,4 Cases have been reported in European, Asian, Middle Eastern, North America, and African countries.5 For the Latin American region, cases have been reported only from Brazil and Chile.6,7,8,9
ARSACS is classically characterized by a triad of early-onset, slowly progressive cerebellar ataxia, pyramidal spasticity, and axonal-demyelinating sensorimotor peripheral neuropathy.10 However, significant clinical variability has been described between patients from different countries and between affected members of the same family.11,12 Clinical phenotypes include disease onset in early adult years, spasticity, pyramidal involvement, dysarthria, distal muscle wasting, foot deformities, truncal ataxia, absence of sensory evoked potentials in the lower limbs, retinal striation, and mitral valve prolapse in some cases. Biochemically, hyperbilirubinemia and impaired pyruvate oxidation have been reported.13
The disease is caused by homozygous or compound heterozygous pathogenic variants in the SACS gene. More than 200 pathogenic or likely pathogenic SACS variants have been identified, which mainly affect exon 10 (as of January 2022, https://clinvarminer.genetics.utah.edu/). The gene is located on chromosome 13q12.12 and encodes the sacsin protein.14 Sacsin is a large 520-kDa 5-domain protein; individual domains act as a giant hub functioning at multiple levels of neurofilament biology.15 Sacsin regulates intermediate filament assembly and dynamics in many neuronal populations, including Purkinje and cortical motor neurons. Sacsin is involved in chaperon activities, microtubule balance, and cell migration.11 Several studies suggest that sacsin may also play an essential role in mitochondrial dynamics related to neurodegeneration.16,17
Historically, the clinical diagnosis of ARSACS has been based on the presence of 3 main clinical signs: ataxia, pyramidal involvement, and axonal neuropathy. However, it is often difficult to establish a diagnosis due to the lack of typical clinical manifestations or the presence of other signs and symptoms such as deafness, intellectual disability, or seizures.18 Differential diagnoses include other spinocerebellar recessive ataxias, such as FA, abetalipoproteinemia, ataxia-telangiectasia, Troyer syndrome, spastic paraplegia 7, autosomal recessive ataxia with vitamin E deficiency, and spastic paraplegia 30. Genetic testing is required for the specific diagnosis of ARSACS.19
Over the past years, massively parallel sequencing for whole-genome, whole-exome, or clinical exome sequencing and the development and application of powerful bioinformatic tools have promoted significant advancements in research and diagnosis of common and rare inherited diseases.20 However, in low- and middle-income countries in Latin America, the use of these methodologies in routine diagnosis is limited by cost. This situation contributes to significant delays in establishing the etiological diagnosis of patients with rare diseases, such as SCAR, and the lack of epidemiological data on the mutational spectrum of most genetic diseases.21
In collaboration with other public health institutions, the Genomic Diagnostic Laboratory from the National Institute of Genomic Medicine has been performing exome sequencing in patients with clinical suspicion of SCAR to identify its genetic cause. This approach allowed us to report the first cases of spastic ataxia of Charlevoix-Saguenay in Mexico and to describe 2 new mutations in the SACS gene. This study expands on the mutational spectrum and geographic location of the disease.
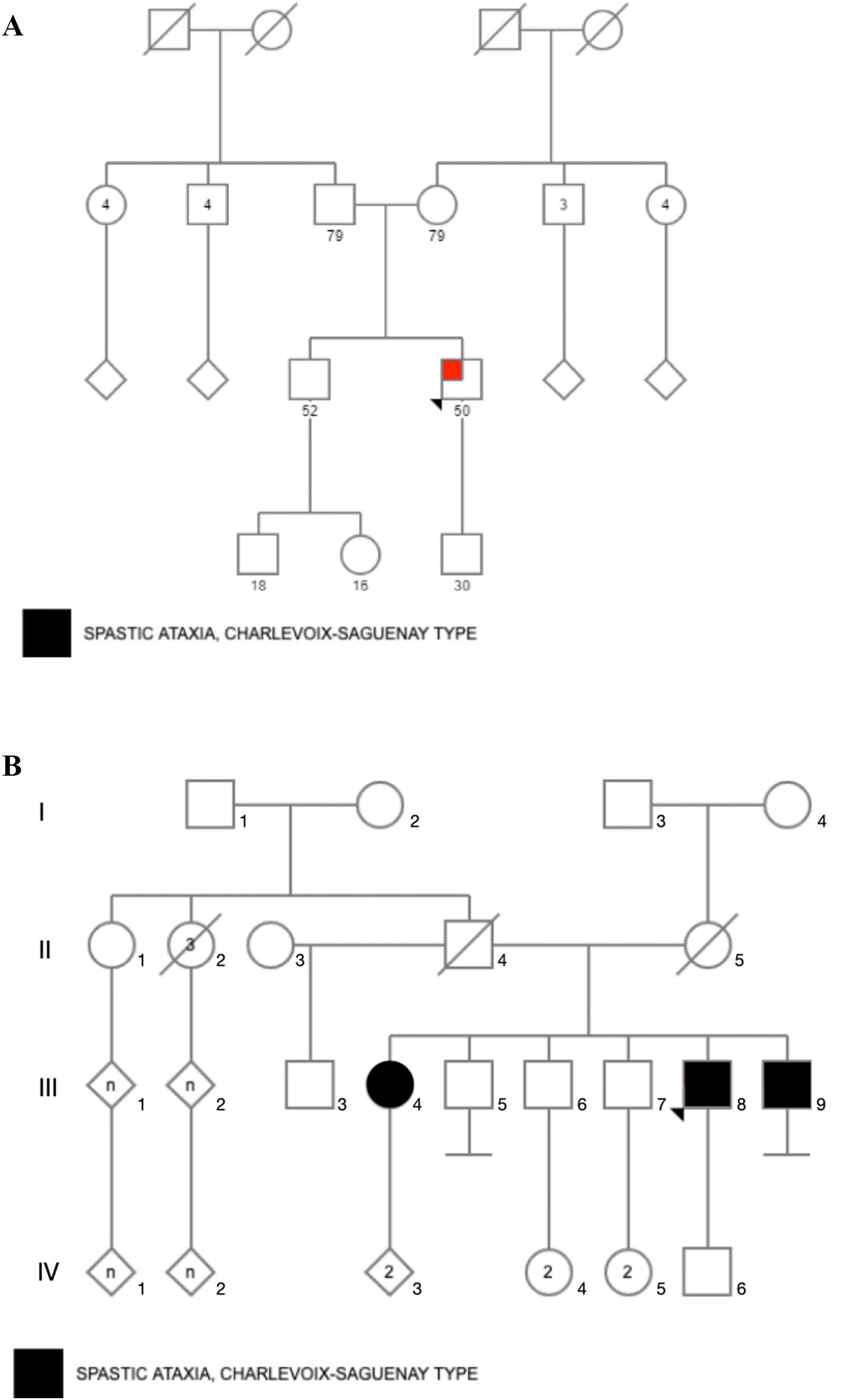
Material and methodsStudy design and participantsFour Mexican mestizo patients of 2 unrelated families (Fig. 1) with clinical suspicion of SCAR were evaluated by clinical geneticists and neurologists. The evaluation confirmed that patients had chronic and progressive gait or limb ataxia. All of them tested negative for FA by PCR analysis.22 Acquired causes of ataxia were also ruled out according to standard protocols.23
. B. Pedigree of family 2 (patients 2, 3, & 4).")
Written informed consent for clinical evaluation, exome sequencing, and data report was obtained from all studied participants. A clinical geneticist provided pre- and post-test counseling to patients and their families. A clinical report with the genetic findings was given to each patient. The National Institute of Genomic Medicine in Mexico provided financial support to conduct this research study.
DNA quality and quantityGenomic DNA (gDNA) was extracted from peripheral blood samples using Maxwell® 16 Blood DNA Purification Kit (Promega, Madison, WI, USA). DNA purity and concentration were determined using NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). A total of 50 ng determined by Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) was used for clinical exome sequencing.
Clinical exome sequencingLibrary preparation was performed according to the manufacturer's protocol using the reagents provided in the Clinical Exome Solution panel kit v2 (Sophia Genetics SA, Saint-Sulpice, Switzerland). Sequencing (2 x 150 bp paired-end reads) was performed on NextSeq Instrument (Illumina, San Diego, CA, USA). Sequencing data analysis and variant annotation were performed using Sophia DDM® (Sophia Genetics SA, Saint-Sulpice, Switzerland), Varsome (https://varsome.com/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Franklin by Genoox (https://franklin.genoox.com/clinical-db/home) platforms. Scientific literature was also consulted for variant annotation and classification. Variants were classified following the criteria published by the American College of Medical Genetics and Genomics.24
FASTQ files were aligned against the reference sequence of the human genome of the National Center for Biotechnology Information of the National Institutes of Health version 37/hg19. Bioinformatic filters were applied, including genes previously reported to be related to SCARs and phenotypes such as ataxia (HP:0001251), peripheral neuropathy (HP:0009830), cerebellar atrophy (HP:0007360), spinal atrophy (HP:0007344), and spasticity (HP:0001257). The coverage depth for variants analyzed ranged from 96 to 135x, with a mean of 115x. The transcript NM_014363.6 was used for SACS gene variant annotation.
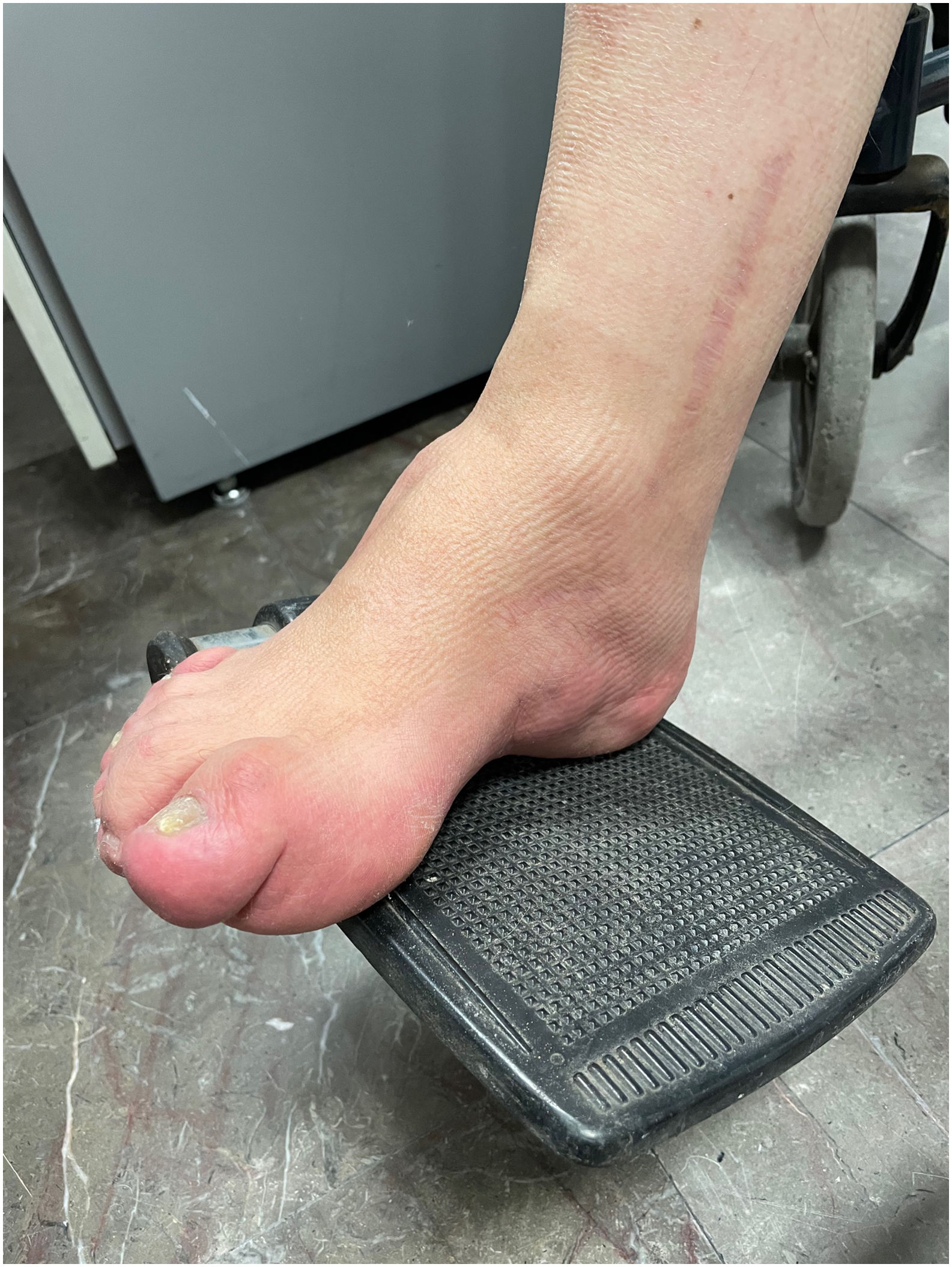
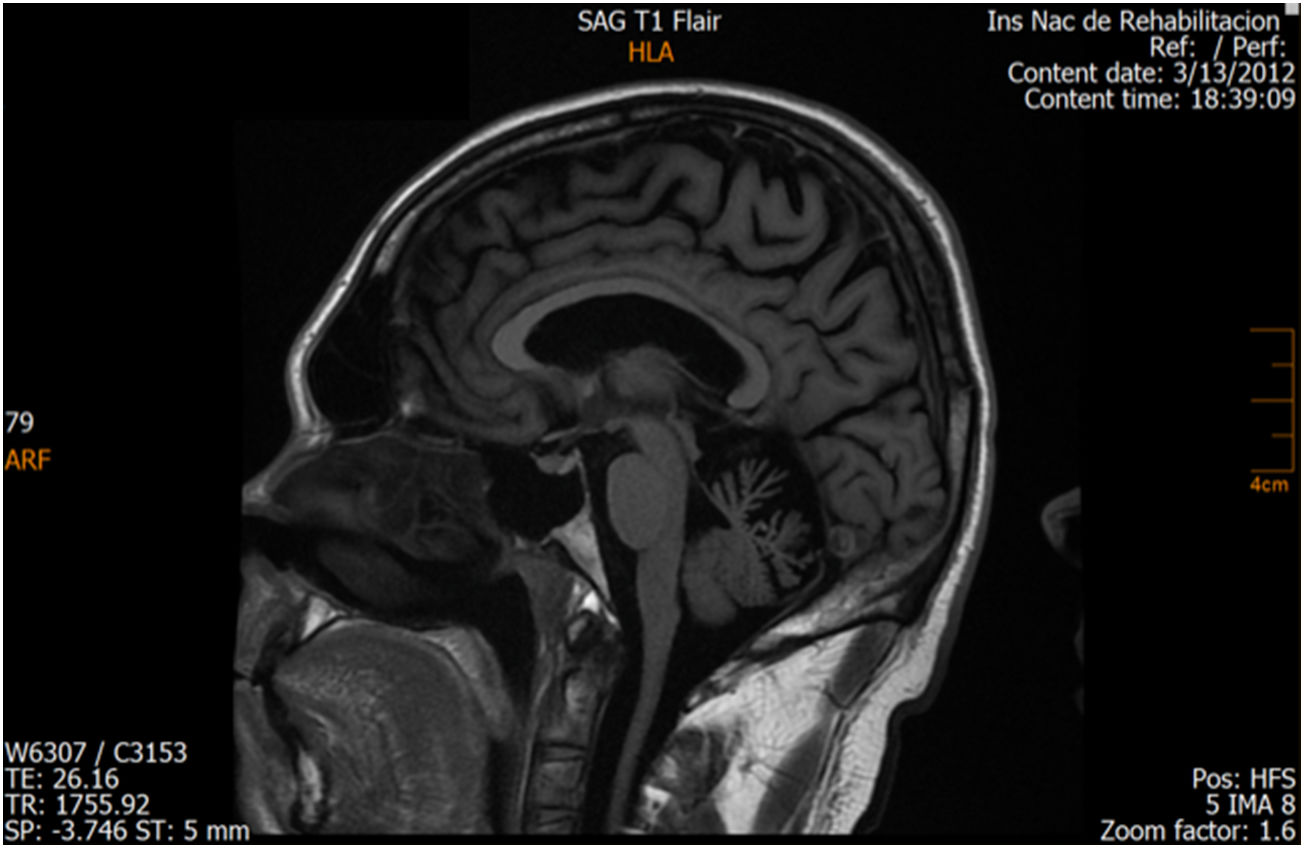
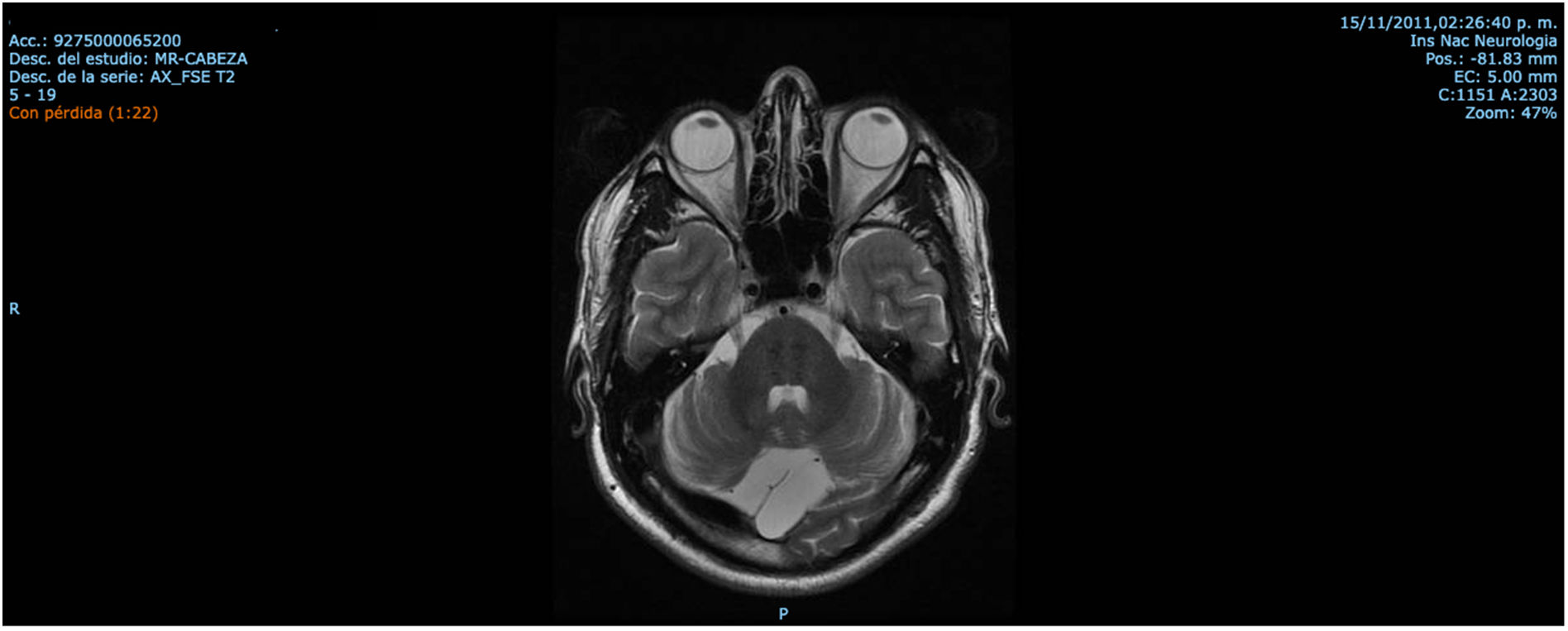
ResultsCase 1 is a 50-year-old male born from a non-consanguineous marriage in Mexico City (Fig. 1A). At age 5, he developed progressive ataxia, dysmetria, cerebellar tremor, and slurred speech. The initial neurological examination revealed bilateral gaze-evoked nystagmus, preserved strength 5/5 except for distal lower limbs (2/5), absent reflexes in all extremities, increased muscle tone in lower limbs, absent Babinski sign, and decreased vibration sensation in lower extremities. Clinical findings are summarized in Table 1. Pes cavus and hammertoes were evident on the last examination at age 47 (Fig. 2); gait was ataxic and only possible with crutches. Brain MRI revealed severe cerebellar and cervical spine atrophy (Fig. 3).
Summary of clinical and imaging findings.
| Gender | Age | AAOa | Clinical manifestations | MRI findings | |
|---|---|---|---|---|---|
| Patient 1 (Family 1) | M | 47 | 5 | Ataxia, slurred speech, nystagmus, spasticity and decreased vibration sensation in lower extremities, pes cavus and hammertoes | Severe cerebellar and cervical spine atrophy Fig. 3 |
| Patient 2 (Family 2) | M | 41 | 30 | Ataxia, tremor, dysarthria, spasticity, pes cavus, mild cognitive impairment, nystagmus, complete pyramidal syndrome, decreased sensitivity in distal extremities | Cerebellar and cortical atrophy, hypointense pontine striae, retrovermian arachnoid cyst Fig. 4 and Fig. 5 |
| Patient 3 (Family 2) | M | 45 | 10 | Ataxia, mild cognitive impairment, spasticity, slurred speech, pyramidal syndrome, decreased sensitivity in distal extremities | Cerebellar and cortical atrophy, retrovermian arachnoid cyst similar to brother Fig. 6 |
| Patient 4 (Family 2) | F | 50 | 15 | Ataxia, slurred speech, mild cognitive impairment, urinary incontinence | Decreased subcortical and supratentorial cortical volume, superior cerebellar vermis atrophy, hypointense pontine striae |


Sequencing results showed the patient was compound heterozygous for the following variants: SACS(NM_014363.6):c.1201C > T(p.Arg401*) and SACS(NM_014363.6):c.11624G > A(p.Arg3875His) (Table 2). Variant c.1201C > T(p.Arg401*) is located in exon 8, which creates a premature translational stop at codon 401. This mutation is expected to result in an absent protein product since 4179 amino acids are lost. According to the Genome Aggregation Database (gnomAD), its frequency is very low; it has only been identified in 1 individual of European ethnicity (as of January 2022). The variant was not found in a local database of 480 Mexican mestizo exomes. Loss-of-function variants in SACS are known to be pathogenic. Additionally, the variant is enlisted in the ClinVar database (Allele ID 840614) and classified as pathogenic (as of January 2022) and is also classified as pathogenic according to the ACMG criteria.24
Variant findings in the SACS gene.
| Gene | cDNA varianta | Exon | Protein change | Domainb | ClinVar variation ID | Type | |
|---|---|---|---|---|---|---|---|
| Patient 1 (Family 1) | SACS | c.1201C > T | 8 | p.Arg401a | SRR1 | 843 602 | Compound heterozygous |
| c.11624G > A | 10 | p.Arg3875His | Between XPCB-DNAJ | 424 796 | |||
| Patients 2, 3 & 4 (Family 2) | SACS | c.475 T > G | 7 | p.Trp1279a | Between SRR1 & SRR2 | Novel | Compound heterozygous |
| c.3836G > A | 10 | p.Tyr159Asp | SRR1 | Novel |
The variant c.11624G > A, originating the missense p.Arg3875His, lies in exon 10. It is absent from exomes and genomes in the gnomAD and from our local database of 480 Mexican mestizo exomes (as of January 2022). The computational prediction for this variant is mainly pathogenic (BayesDel_addAF, DANN, EIGEN, FATHMM-MKL, LIST-S2, M-CAP, MVP, MutationTaster, and PrimateAI algorithms). Only DEOGEN2, MutationAssessor, and SIFT predicted it as benign. This variant has been previously reported in a patient with the disease.25 According to the available information, the variant is classified as likely pathogenic.
Segregation of the variants in the healthy parents confirmed that variant p.Arg3875His was inherited from the father and p.Arg401Ter from the mother. The exome sequencing results established the diagnosis of ARSACS in this patient.
Cases 2, 3, and 4 belong to the same family; they were born from a non-consanguineous marriage (Fig. 1B). Both parents were from Guanajuato, and the 3 siblings were born in the State of Mexico.
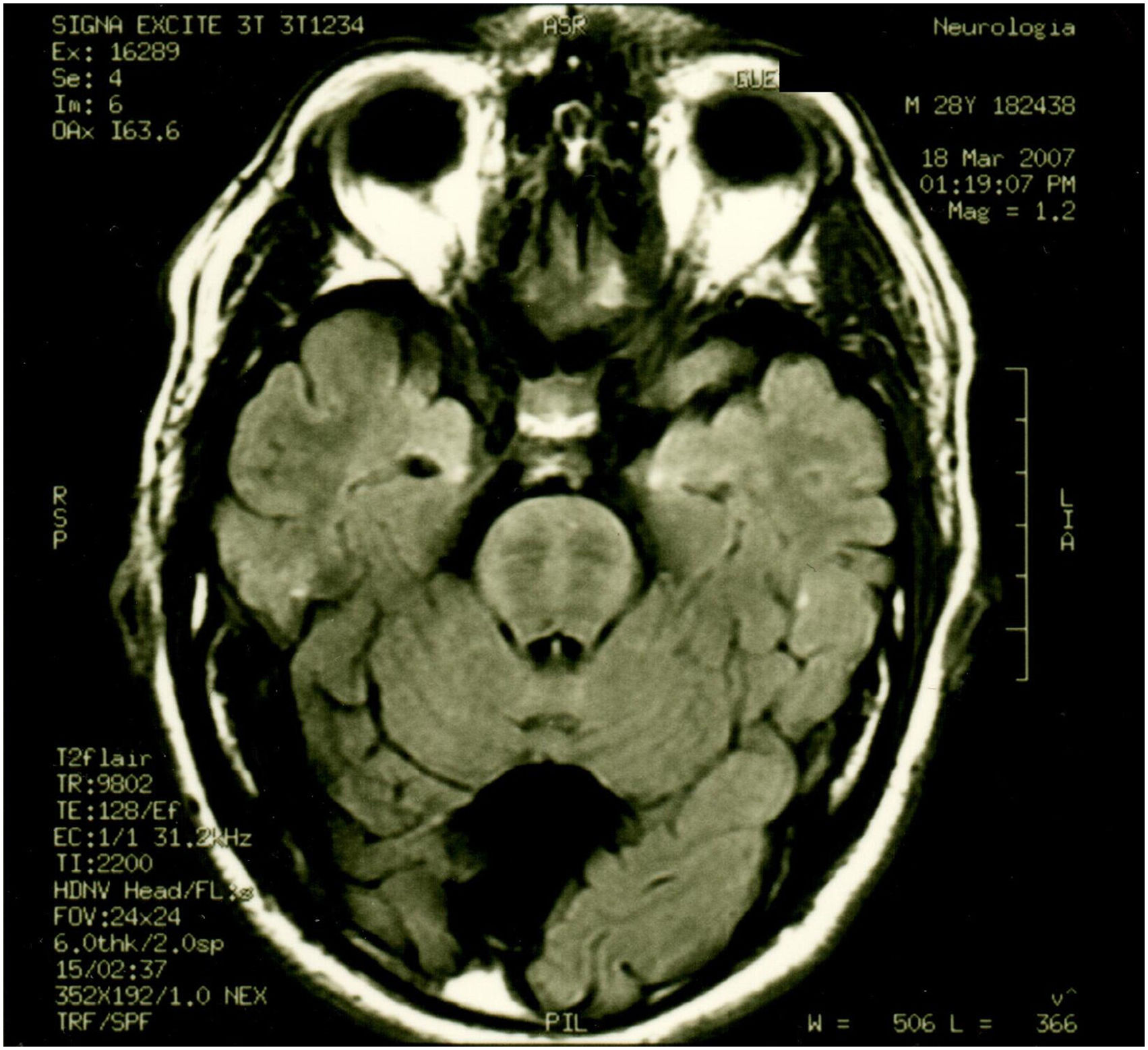
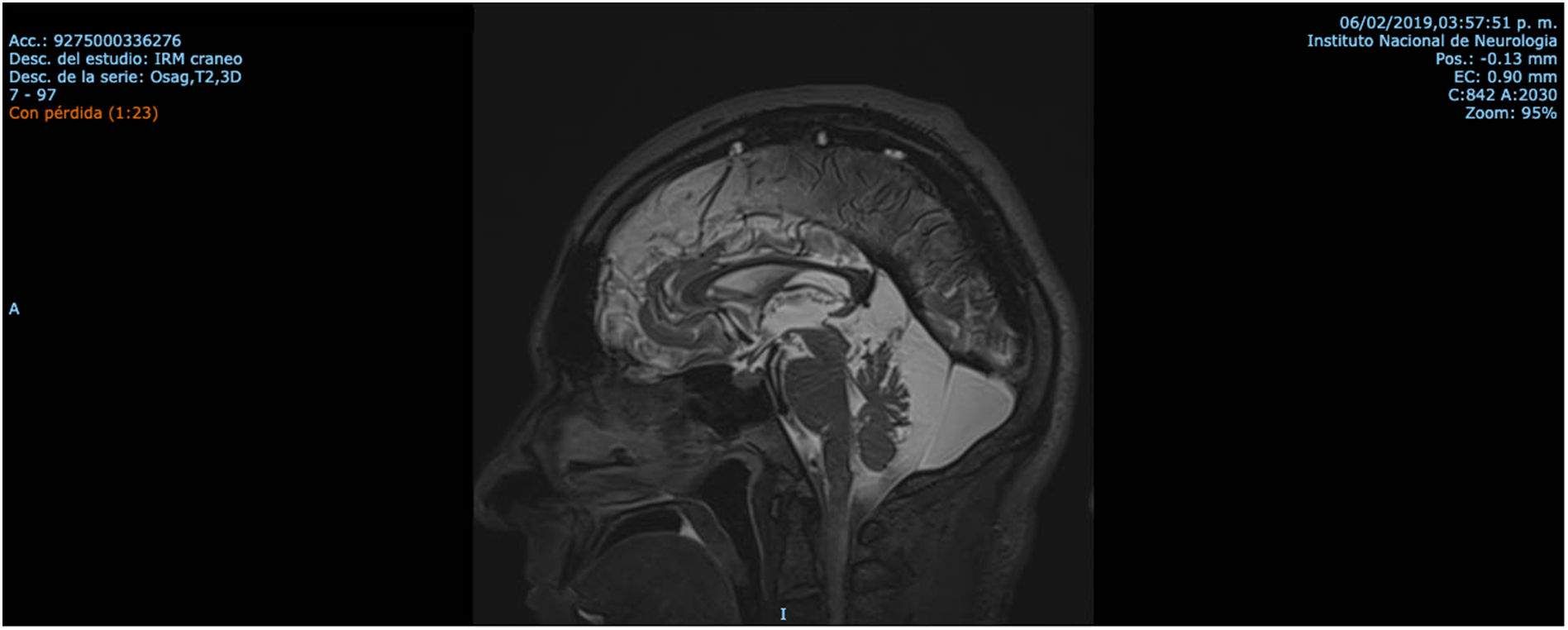
Patient 2 is the index case; he is a 41-year-old male with 5 siblings, 2 of whom have ataxia (Patients 3 and 4 in this report). Symptoms began at age 30; he had recurrent falls due to balance alteration and indistinct lateralized gait, clumsiness to grasp things with his left hand, nasal voice, progressive dysarthria, and spasticity. Mild cognitive impairment and bilateral horizontal nystagmus with slow saccades were found on physical examination (Table 1). MRI revealed decreased cortico-subcortical volume, a retrovermian arachnoid cyst, cerebellar atrophy, and typical hypointense pontine striae (Fig. 4 and Fig. 5).


Patient 3 is a 45-year-old male who noticed unsteadiness and frequent falls from age 10. His symptoms were progressive, and he used a walking aid. On examination, his speech was slurred but intelligible. Hoffman and Trömner signs and exalted patellar reflexes, and spasticity of the lower limbs were present. Bilateral Achilles reflexes were abolished. The patient complained of leg paresthesia, numbness, and cramps. Abolition of deep sensitivity was evident from the knee downwards (Table 1). MRI findings (Fig. 6) were similar to his brother's (Patient 2).

Patient 4 is a 50-year-old woman with progressive gait ataxia, slurred speech, and mild memory impairment at age 15. Physical findings were very similar to those of her siblings. MRI revealed decreased subcortical and supratentorial cortical volume. Discrete supratentorial leukopathies plus dilated Virchow's spaces in basal ganglia were evident. In T2-weighted and FLAIR images, atrophy of the superior vermis and hypointense striae at the pontine level were observed (Table 1).
Sequencing results for the index case (Table 2) revealed 2 new variants in the SACS gene: NM_014363.6:c.3836G > A(p.Trp1279*) located in exon 10 and NM_014363.6:c.475 T > G(p.Tyr159Asp) located in exon 7. p.Trp1279* produces a premature stop codon that is predicted to cause the loss of 3301 amino acids in the protein. Population frequency is very low; only 1 carrier was identified in the ExAC database (March 2022). It is absent from exomes and genomes of the gnomAD and exomes of 480 Mexican individuals. Since loss-of-function is a known disease mechanism, we classified p.Trp1279* as pathogenic.
p.Tyr159Asp is predicted to be deleterious by BayesDel_addAF, DANN, DEOGEN2, EIGEN, FATHMM-MKL, LIST-S2, M-CAP, MVP, MutationAssessor, MutationTaster, and SIFT. It is absent from exomes and genomes of the gnomAD and from the local database of 480 Mexican mestizo exomes (as of January 2022). Considering that the variant is found in trans with a pathogenic variant in SACS and that both variants were identified in the other 2 affected siblings (Patients 3 & 4), we classified this variant as likely pathogenic. This variant is not present in the ClinVar database (as of January 2022).
DiscussionWe report the mutations in the SACS gene in 4 Mexican individuals with cerebellar ataxia for the first time. Thus, the current geographic location of the disease and the mutational spectrum of the SACS gene are expanded. Several cases of ARSACS may still be unidentified in Latin America due to limited access to molecular testing for most patients in this region.
Many phenotypes for spastic ataxia of Charlevoix-Saguenay have been described, ranging from early-onset ataxia with spasticity to late-onset ataxia without it. Dysarthria, nystagmus, pyramidal tract signs, sensory-motor peripheral neuropathy, hypermyelination of retinal fibers, and atrophy of the superior cerebellar vermis have been identified as additional but atypical features.26 More epidemiological information on the frequency of these other signs and symptoms and their onset in the disease progression are still needed.
Patients with ARSACS usually exhibit early onset and progression of spasticity in the lower limbs that become apparent between the first and second years of age when they begin to walk.10 In 3 of the 4 patients in our report, difficulty walking due to lower limb spasticity or ataxia was noticed before age 15 but not in the first 2 years. Regardless of their origin, dysarthria and slurred speech have been reported in all ARSACS patients.27 Moreover, pyramidal tract involvement with the Babinski sign is consistently positive in nearly all patients diagnosed with ARSACS, as observed in all our patients.
Some patients show skeletal abnormalities, such as hammertoes and pes cavus. These abnormalities were evident during the examination of Patient 1. Musculoskeletal changes have been well established in Charcot–Marie–Tooth disease and other neurodegenerative diseases including ARSACS.28,29
Mild cognitive impairment was detected in all patients from the same family. Some of the cognitive and affective deficits observed in ARSACS have been postulated to form the so-called cerebellar cognitive, affective syndrome (CCAS). Lesions of the posterior lobe and the cerebellar vermis have been proposed to be responsible for this syndrome.18,30 Intellectual disability has been found in 11.4% of patients with ARSACS; the most common findings are ataxia (78.9%), spasticity (78.1%), and polyneuropathy (73.7%).5 Our patients consistently showed a uniform and recognizable clinical phenotype characterized by cerebellar ataxia, lower limb spasticity, sensorimotor axonal neuropathy, and cerebellar and cervical spine atrophy on brain MRI. Mitral valve prolapse was initially reported as a frequent abnormality in patients diagnosed with ARSACS1; data from our patients did not confirm cardiac involvement.
Mexican patients with ARSACS exhibit the same MRI findings previously reported in patients from Canada, Brazil, and Europe. Irrespective of genotype, MRI findings revealed cerebellar atrophy and mild atrophy of the cervical spinal cord, and linear hypointensities in the pontine parenchyma akin to previous reports.6,31,32
Intra- and interfamilial clinical heterogeneities have been observed in affected individuals sharing the same mutations.12,13,33 The age at onset of the index case was 30 years, while in the other 2 affected siblings, symptoms started during infancy.
It has been suggested that genetic or environmental factors could impact disease phenotype. This idea is supported by recent findings showing that sacsin is almost absent from ARSACS fibroblasts, regardless of the nature and position of the mutation along the gene. These findings suggest that phenotypic variability in ARSACS is not due to different levels of residual sacsin or positional effect of the mutations and indicate that lack of sacsin is the pathogenic mechanism shared by other SACS mutations.34 Further investigation is needed to identify determinant factors that act as phenotype modifiers.
Cases of autosomal recessive spastic ataxia of Charlevoix-Saguenay have been described in Canada (Quebec), the United States, Brazil, Chile, Tunisia, Turkey, Japan, Italy, Spain, the Netherlands, Belgium, Russia, and Germany.35,36 Bi-allelic mutations in the SACS gene cause ARSACS; more than 200 pathogenic single nucleotide variants, mostly missense type, have been reported in the literature and specialized databases.37 With the increasing adoption of exome sequencing to diagnose different forms of inherited ataxia or spastic paraplegia worldwide, reported mutations are rapidly growing.
This study reports 2 new disease-causing variants in the SACS gene. Both variants segregated with the disease in all patients of the same family. Both variants have already been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) (Submission-ID: SUB11168262, Organization ID: 507106). No further functional or in vitro analyses were performed to clarify the pathogenicity mechanism for each variant. However, p.Trp1279* is predicted to cause a loss of function due to an early stop codon. In vitro analysis of ARSACS cells carrying truncating mutations in the SACS gene show a significant reduction of SACS mRNA compared with controls,34 which suggests that a nonsense-mediated decay mechanism degrades mRNA. The mRNA degradation causes loss of function related to the pathogenicity of truncating variants. It is possible that this mechanism could be acting for p.Trp1279*.
p.Tyr159Asp, a missense likely pathogenic new variant, is located in the sacsin repeating region 1. Missense mutations in this region may result in lower stability and abnormal conformation of sacsin.11 Analyses of other SACS missense mutations have revealed that the sacsin polypeptide undergoes a cotranslational quality control leading to the ubiquitination and degradation of nascent proteins that cannot fold correctly. Ubiquitinated degradation products of nascent mutant sacsin have been identified in ARSACS patient cells carrying missense mutations.34 This mechanism is predicted to occur for large and multimodular proteins, whose folding occurs cotranslationally.38
ARSACS patients can exhibit symptoms similar to those of other SCAR forms, thus making it difficult to establish a precise etiologic diagnosis. Genetic testing should always be conducted in patients with clinical suspicion of SCAR to identify the causal genetic variants and offer genetic counseling to relatives. There are no specific clinical tests with high diagnostic yield useful for diagnosing SCAR, especially within such a diverse patient population with very high phenotypic and genetic heterogeneity. Most of the diagnostic results are negative, and the diagnostic workup can take many years.
Therefore, next-generation exome sequencing with CNV prediction is a more cost-effective methodology to diagnose patients with rare diseases, including ataxia. This approach allows a broader range of genes to be tested than gene panels, including genes associated with other neurologic disorders besides ataxia-related genes. The genomic approach reduces the time to diagnosis and shortens the “diagnostic odyssey.” It often avoids unnecessary and non-specific tests and reduces overall costs. Moreover, in cases where a specific treatment is available, it can be started immediately to benefit the patient.
The overall diagnostic yield using next-generation sequencing methods was around 35% for rare inherited diseases likely to have Mendelian inheritance and a single-gene etiologic factor. According to recently published data, most patients in this group had an intellectual disability, neurological, or neurodevelopmental disorders in the foreground.39 In the past decade, a significant amount of data has been generated using different sequencing methods. New sequencing methods and technologies will eventually supplant the current ones, but the collected data will continue to be used in collaborative SCARs research.
All participants gave their informed consent prior to their inclusion in this study.
Conflict of interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial disclosureThe National Institute of Genomic Medicine in Mexico provided financial support to conduct this research study.
Informed consentThe authors declare and guarantee that they are in possession of a document signed by the patients authorizing the inclusion of their data in the article and its subsequent publication, distribution and circulation.
