




Nontuberculous mycobacteria (NTM) is characterized by refractory infection that often results in structural destruction of the lungs and respiratory failure, making its management challenging. Bronchiectasis often develops over the course of the NTM lung disease (NTM-LD). A microbiome analysis can give detailed bacterial flora information within the bronchoalveolar region that cannot be obtained using conventional culture tests. Previous reports have indicated that the microbiome composition is closely linked to the characteristics and pathology of each respiratory disease.1–4 Although there have been several previous studies in patients with NTM-LD, the microbiome compositions reported have varied,3,5 so more microbiome datasets from various centers that show the heterogeneity of clinical outcomes in patients with NTM-LD are needed. In this study, we analyzed the microbiome in order to investigate differences between these two conditions.
We prospectively recruited patients with suspected NTM-LD who were undiagnosed from sputum culture and had been referred for bronchoscopy. Patients with a diagnosis of NTM confirmed by bronchoscopy were included in the NTM group and those without NTM were included in the bronchiectasis group. Bronchoalveolar lavage fluid (BALF) was collected from all patients using bronchoscopy, with portions stored at −80°C for research. In this study, stored BALF samples were used. Details of microbiome analysis are provided in Supplementary File 1. This study was approved by the Institutional Review Board of the National Hospital Organization Kyoto Medical Center (approval number: 16-040) and the study protocol was registered in the UMIN Clinical Registry (Number: UMIN000027652).
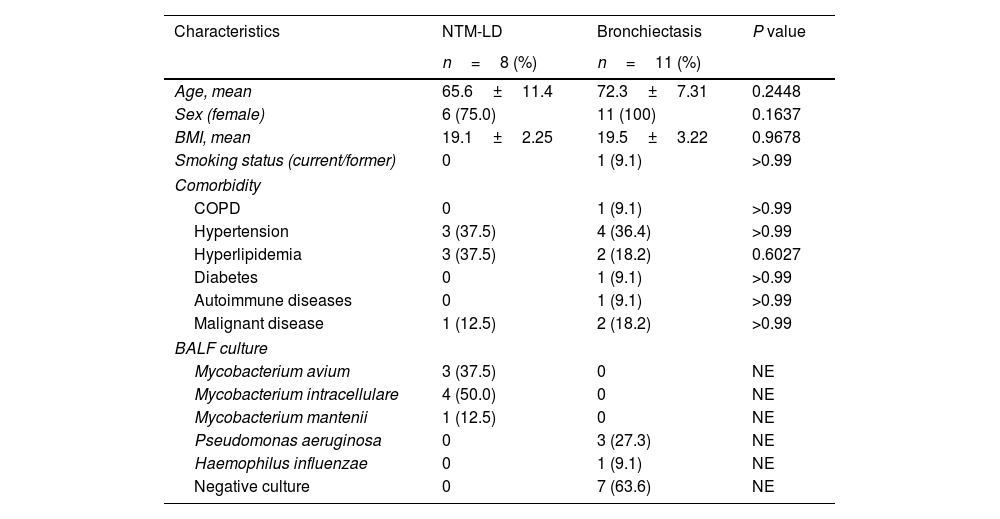
Nineteen patients were recruited for this exploratory study. Following bronchoscopy procedures, eight (42.1%) patients were diagnosed with NTM-LD, and 11 (57.9%) with bronchiectasis without NTM. Diagnostic criteria from the American Thoracic Society/Infectious Disease Society of America guidelines were used to diagnose NTM.6Table 1 shows the characteristics of study participants, who were predominantly female (89.5%) with a mean age at bronchoscopy of 69.5±9.3 years. The species found to be responsible for NTM-LD were Mycobacterium avium in three cases, Mycobacterium intarcellulare in four cases, and Mycobacterium mantenii in one case. In patients with bronchiectasis, the strain identified in three cases was Pseudomonas aeruginosa and one case Haemophilus influenzae. Seven samples from patients with bronchiectasis were culture-negative.
Characteristics of patients with NTM-LD and bronchiectasis.
| Characteristics | NTM-LD | Bronchiectasis | P value |
|---|---|---|---|
| n=8 (%) | n=11 (%) | ||
| Age, mean | 65.6±11.4 | 72.3±7.31 | 0.2448 |
| Sex (female) | 6 (75.0) | 11 (100) | 0.1637 |
| BMI, mean | 19.1±2.25 | 19.5±3.22 | 0.9678 |
| Smoking status (current/former) | 0 | 1 (9.1) | >0.99 |
| Comorbidity | |||
| COPD | 0 | 1 (9.1) | >0.99 |
| Hypertension | 3 (37.5) | 4 (36.4) | >0.99 |
| Hyperlipidemia | 3 (37.5) | 2 (18.2) | 0.6027 |
| Diabetes | 0 | 1 (9.1) | >0.99 |
| Autoimmune diseases | 0 | 1 (9.1) | >0.99 |
| Malignant disease | 1 (12.5) | 2 (18.2) | >0.99 |
| BALF culture | |||
| Mycobacterium avium | 3 (37.5) | 0 | NE |
| Mycobacterium intracellulare | 4 (50.0) | 0 | NE |
| Mycobacterium mantenii | 1 (12.5) | 0 | NE |
| Pseudomonas aeruginosa | 0 | 3 (27.3) | NE |
| Haemophilus influenzae | 0 | 1 (9.1) | NE |
| Negative culture | 0 | 7 (63.6) | NE |
Abbreviations: BALF, bronchoalveolar lavage fluid; BMI, body mass index; COPD, chronic obstructive pulmonary disease; NTM-LD, nontuberculous mycobacterial lung disease.
Fig. 1 shows the microbiome differences in terms of alpha and beta diversities between the NTM-LD and bronchiectasis groups. Fig. 1A–C shows the Chao1 (H=1.970, P=0.160), Simpson (H=5.734, P=0.0166), and Faith's phylogenetic diversity indexes (H=1.371, P=0.242), respectively. There were no differences between the NTM-LD and bronchiectasis groups in terms of Chao1 and Faith's phylogenetic diversity indexes; however, patients with NTM-LD were found to have significantly higher Simpson indexes than those with bronchiectasis. Fig. 1D and E shows the Bray–Curtis (P=0.016) and weighted UniFrac distances (P=0.002) used to evaluate beta diversity. There were significant differences in terms of beta diversity between the patients with NTM-LD and those with bronchiectasis.
 Chao1 index; (B) Simpson index; and (C) Faith")
 Chao1 index; (B) Simpson index; and (C) Faith")
Calculation of alpha diversity values. (A) Chao1 index; (B) Simpson index; and (C) Faith's phylogenetic diversity index. Calculation of beta diversity values. (D) Bray–Curtis distance; (E) weighted UniFrac distance. Red dots indicate NTM and blue dots indicate bronchiectasis. Fig. (F) and (G) shows the taxa bar plots of patients with NTM-LD (F) and bronchiectasis (G). Fig. (H) shows a heat map of the relative abundance of the major bacteria in both groups of patients.
Fig. 1F and G shows the taxon bar plots of patients with NTM-LD and bronchiectasis. Our microbiome analysis revealed that Streptococcus, Prevotella, and Staphylococcus were most abundant in the NTM-LD group, and Pseudomonas, Haemophilus, and Staphylococcus were predominantly present in the bronchiectasis group. The bronchiectasis group was often dominated by a few genera, whereas the NTM-LD group included several genera that were less frequently detected. The top 10 taxa for each sample are listed in Supplementary Table 1. Notably, the microbiome of the NTM-LD group was mainly characterized by three genera – Streptococcus, Prevotella, and Rothia – in 5/8 (62.5%) of the samples. We also found that the bacteria in the BALF cultures from both groups were not always present in the microbiome analysis, particularly in 5/8 of the patients with NTM-LD, where NTM strains were not detected. Fig. 1H shows a heat map of the relative abundance of the major bacteria in both patient groups. The heat map shows a clear division of bacterial flora into two clusters: NTM-LD and bronchiectasis.
This study showed differences in microbiome diversity in BALF samples collected from patients with NTM-LD and bronchiectasis. The microbiota of patients with NTM-LD was found to be more diverse than that of patients with bronchiectasis. Notably, the bacterial flora in the bronchoalveolar area was highly diverse in patients with NTM infection.
Significant differences were also found in the composition of the bacterial flora. Patients with bronchiectasis showed a dominance of specific genera, each accounting for >50% of the total, while patients with NTM-LD showed fewer dominant genera, leading to a more evenly distributed overall genera diversity. In previous reports,3,5Haemophilus, Pseudomonas, and Streptococcus were reported to be the most frequently detected genera in patients with bronchiectasis, whereas Streptococcus, Prevotella, Veillonella, and other genera were reported to be more common in patients with MAC-LD. Although similar genera were detected in all the BALF samples in our study, the compositional proportions were found to differ significantly. In patients with bronchiectasis, a small number of specific genera made up the majority of the flora, resulting in less diversity. In contrast, in patients with NTM-LD, the flora was composed of a larger variety of genera, resulting in a high level of diversity. This was also supported by the two clusters clearly shown on the heatmap. A recent study from South Korea reported that patients with NTM-LD have less microbiome diversity in sputum compared to the general healthy population.7 The difference with our study is that the analysed samples were sputum and BALF, and the analysed controls were healthy subjects and bronchiectasis patients, so a simple comparison cannot be made; nevertheless, the results are interesting. Compared to the general healthy population, the microbiome diversity of patients with NTM-LD may be reduced. We also identified several characteristic genera found in the bacterial flora of patients with NTM-LD. These include Streptococcus, Prevotella, and Rothia – all of which are endemic to the oral cavity and can cause serious infections in immunocompromised individuals. These three bacteria are also known biomarkers of lower airway inflammation.8 This is consistent with reports that anaerobic bacteria, mainly of the genus Prevotella, are frequently detected in patients with NTM infection.9 Patients with NTM-LD often develop coinfection with other bacteria, such as Staphylococcus aureus, P. aeruginosa, and H. influenzae.10 Although certain strains of bacteria account for a greater proportion of disease, it has been suggested that the diversity of these organisms is maintained in the early stages of diagnosis.
In addition, as previously reported, many of the NTM bacteria detected in the BALF cultures were not detected in our microbiome analysis. This is likely because the 16S rRNA gene sequencing approach is not sensitive enough to identify NTM in airway samples.3,5 Our study also confirms the limited sensitivity of culture-independent methods. This is important because microbiome analysis alone is insufficient for diagnosing active infectious diseases. There were several limitations in this study. It was performed in a single center and may be prone to patient selection bias. In addition, the small number of patients analyzed and the exploratory nature of the study means that the results are unlikely to be applicable directly to other centers.
In conclusion, we found that the lung microbiome of patients with bronchiectasis showed less diversity, whereas that of patients with NTM-LD maintained a higher level of diversity. Although dysbiosis occurs in patients with bronchiectasis, it can be hypothesized that NTM influences disease formation while maintaining microbiome diversity in patients with NTM-LD. Further large-scale studies are warranted to fully clarify the nature of this phenomenon.
FundingNone to declare.
Authors’ contributionsKohei Fujita and Yuki Yamamoto designed this study, collected patient specimens, analyzed and interpreted the data, and wrote and revised the manuscript.
Masahiro Kamita, Kosei Tanaka, and Takeru Nakabayashi planned and designed the study, as well as performed the experiments, analyzed and interpreted the data, and revised the manuscript.
Takuma Imakita, Issei Oi, Osamu Kanai, and Tadashi Mio collected the patient specimens, as well as analyzed and interpreted the data.
Kiminobu Tanizawa and Yutaka Ito planned and designed this study, as well as analyzed and interpreted the data.
Conflicts of interestKohei Fujita has received research funds under contracts from MSD, AstraZeneca, and AN2 Therapeutics outside of the submitted work.
Osamu Kanai has received research funds under contract from AstraZeneca outside the submitting work.
Yuki Yamamoto is a board member and a stockholder of HiLung, Inc.
Masahiro Kamita, Kosei Tanaka, and Takeru Nakabayashi are employees of H.U. Group Research Institute G.K.
All the other authors have no conflicts of interest to declare.
The following are the supplementary data to this article:
