










Supplement “Pulmonary Interstitial Pathology”
More infoIdiopathic pulmonary fibrosis (IPF) is the most common fibrosing lung disease. It is associated with a very poor prognosis. Treatments can delay the progression of IPF, so early diagnosis is fundamental. Radiologists play a fundamental role in the evaluation and accurate diagnosis of IPF. Identifying the characteristic patterns of IPF on high-resolution computed tomography (HRCT) is key in the process of multidisciplinary diagnosis, often obviating the need for surgical lung biopsies. This review describes and illustrates the clinical and imaging findings in IPF in the context of the most recent international guidelines, as well as the differential diagnosis and the role of HRCT in follow-up and assessment of complications.
La fibrosis pulmonar idiopática (FPI) es la enfermedad pulmonar fibrosante más frecuente y se asocia con un pronóstico muy pobre, existiendo actualmente tratamientos para retardar su progresión, lo que hace fundamental su diagnóstico temprano. Los radiólogos tienen un papel fundamental en la evaluación y el diagnóstico preciso de la FPI. La identificación de los patrones radiológicos en la tomografía computarizada de alta resolución (TCAR) es clave en el proceso de diagnóstico multidisciplinar y, con frecuencia, obvia la necesidad de una biopsia pulmonar quirúrgica. En esta revisión, describimos las características clínicas y de imagen de la FPI en el contexto de las guías internacionales más recientes, así como el diagnóstico diferencial, el papel de la TCAR en el seguimiento y las complicaciones.
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive fibrosing interstitial pneumonia of unknown cause associated with the radiological and histological features of usual interstitial pneumonia (UIP).1 It is part of the group of chronic fibrosing idiopathic interstitial pneumonia, along with non-specific interstitial pneumonia (NSIP), from the consensus classification by the American Thoracic Society-European Respiratory Society (ATS-ERS) in 2013.2
IPF is a disease that mainly affects older adults, generally in their 50s and 60s, and is twice as common in men. Risk factors include advanced age, family history, smoking, gastroesophageal reflux, and some environmental exposures.3–5 The prognosis of IPF is poor, with an average survival after diagnosis of three to four years. However, the evolution of IPF can be variable, and while most patients progress rapidly, others are stable or progress more slowly, with prolonged survival times of more than a decade.3,4
Typical clinical manifestations are non-specific and include chronic progressive dyspnoea, dry cough, fatigue, bibasilar dry inspiratory crackles on physical examination, and nail clubbing.1,3 Respiratory function tests generally show restriction with a decreased forced vital capacity, decreased lung volumes, and reduced CO diffusing capacity.3–5
The diagnosis of IPF, as in other diffuse infiltrative lung diseases (DILDs), is based on multidisciplinary assessment of clinical, radiological, and histological data. For this, there are multidisciplinary clinical practice guidelines. The first ATS-ERS guidelines on IPF published date from the year 2000.6 However, it is in the 2011 update of the ATS guidelines, the ERS, the Japanese Respiratory Society and the Latin American Thoracic Association (ATS/ERS/JRS/ALAT)1 when the diagnosis by high-resolution computed tomography (HRCT) becomes more relevant, and radiologists start to play a vital role in the diagnosis and management of this disease. These ATS/ERS/JRS/ALAT guidelines were updated in 2018,3 the same year that the White Paper from the Fleischner Society was published4 on the diagnostic criteria of IPF, and it has been updated again very recently, in May 2022, also including the new concept of “progressive pulmonary fibrosis”.7
HRCT plays a fundamental role in the diagnosis of this pathology, and sometimes makes it possible to dispense with histology in the diagnostic algorithm, by identifying the pattern of UIP or probable UIP in fibrosing interstitial lung disease. Early diagnosis of IPF is particularly important due to the poor prognosis of the disease, allowing for early initiation of antifibrotic treatment that can reduce the deterioration of lung function.3,4,8,9
In this article we will review the role of radiology, especially HRCT, in the management of IPF.
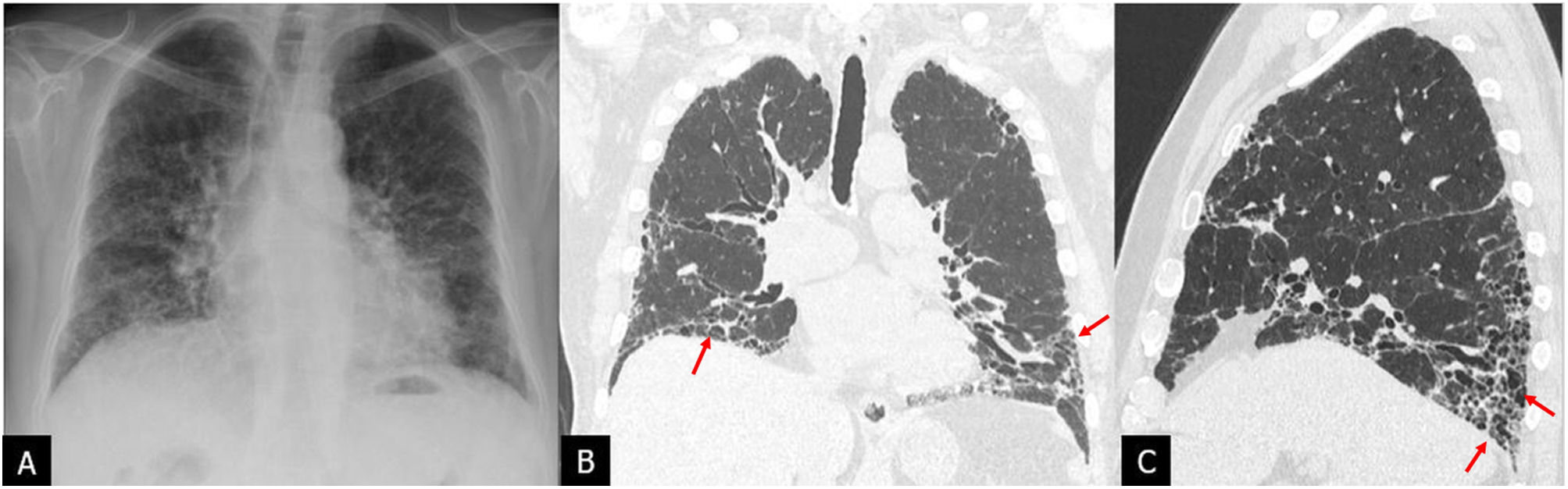
Radiological findings in IPFChest radiograph is, on many occasions, the initial test that is carried out when there is a clinical suspicion of DILD. Chest radiograph findings are non-specific and do not enable accurate characterisation of interstitial lung disease. Pulmonary fibrosis on chest radiograph manifests as reticulation and linear opacities, predominantly basal, and frequently with loss of lung volume in cases of advanced fibrosis10 (Fig. 1A). The current role of chest radiography in DILD is fundamentally to indicate HRCT in the presence of an image showing suspected interstitial involvement for the early diagnosis of IPF. HRCT with the volumetric acquisition (Fig. 1B and C) is the technique of choice for the radiological assessment of IPF and constitutes the first step in its diagnosis. Volumetric HRCT with multi-detector CT provides information about the entire lung, with high-quality images and resolution similar to slices of gross pathology specimens. Acquisition with the appropriate high-resolution technique is essential, in the supine position, with deep inspiration and without intravenous contrast. Acquisition in expiration to demonstrate air trapping is optional, and so is acquisition in the prone position to assess doubtful peripheral interstitial involvement in dependent lung areas.3,4,11,12 Both additional acquisitions can be done low-dose and sequentially.13
 and high-resolution computed tomography (HRCT) with the volumetric acquisition in coronal (B) and sagittal (C) planes in a patient with idiopathic pulmonary fibrosis. The chest radiograph reveals peripheral reticular opacities, predominantly basal, which are indicative of interstitial lung disease, without characterisation of the pattern. HRCT reveals a pattern of usual interstitial pneumonia with traction bronchiolectasis and images of predominantly basal subpleural honeycombing (arrows).")
Chest radiograph (A) and high-resolution computed tomography (HRCT) with the volumetric acquisition in coronal (B) and sagittal (C) planes in a patient with idiopathic pulmonary fibrosis. The chest radiograph reveals peripheral reticular opacities, predominantly basal, which are indicative of interstitial lung disease, without characterisation of the pattern. HRCT reveals a pattern of usual interstitial pneumonia with traction bronchiolectasis and images of predominantly basal subpleural honeycombing (arrows).
The radiological findings present in IPF that characterise the UIP patterns are defined by their subpleural and predominantly basal distribution: honeycombing, traction bronchiectasis, and reticular and ground-glass opacities. These findings have been described in a previous chapter of the supplement, and we will only highlight a few points.
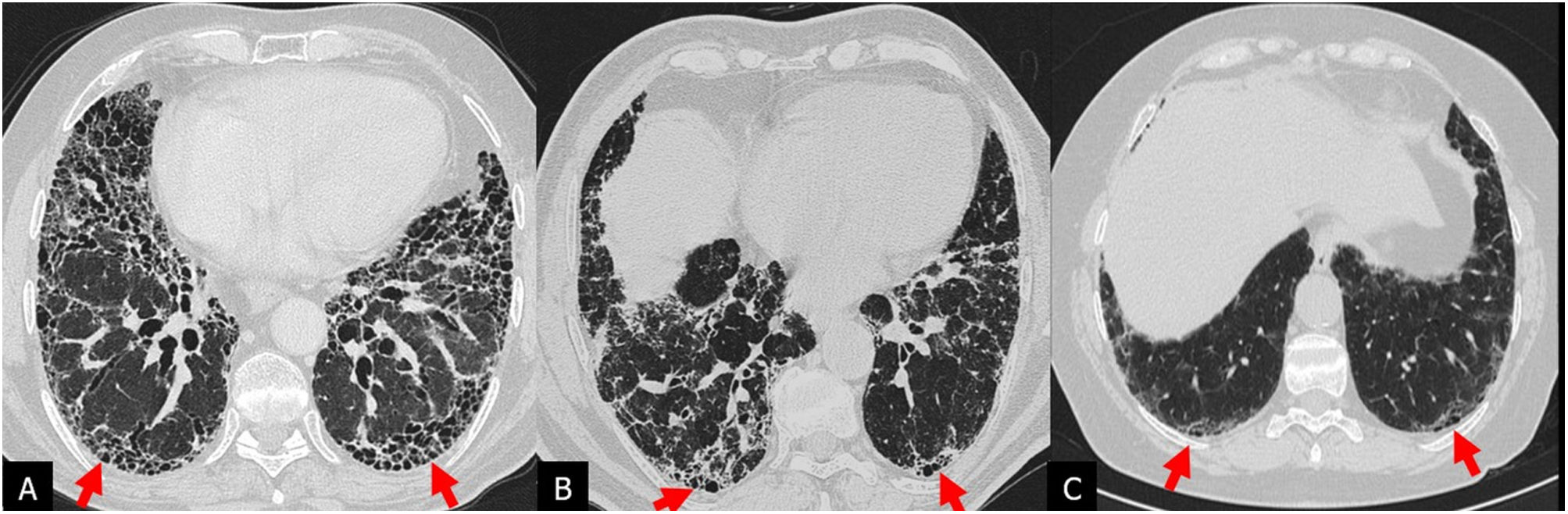
Honeycombing: its identification is key and essential for the diagnosis of the typical UIP pattern. Although honeycombing typically consists of usually subpleural air cysts, 3–10mm in diameter and occasionally up to 2.5cm, grouped and forming layers.14 a single subpleural layer of two or three contiguous cysts is sufficient to diagnose honeycombing.15 The identification of honeycombing can be difficult when subpleural cysts are small and scanty, sometimes requiring additional CT acquisition in the prone position12 (Fig. 2). The two most typical differential findings that mimic honeycombing are paraseptal emphysema and traction bronchiolectasis. Given the difficulty in interpreting the honeycombing, the interobserver variability is moderate.16,17
 images in the axial plane (A and B) reveal inconclusive posterior basal subpleural honeycombing images. Additional HRCT acquisition in the prone position (C and D) confirms the presence of honeycombing (arrows).")
Pattern of usual interstitial pneumonia in a patient with idiopathic pulmonary fibrosis. Inspiratory high-resolution computed tomography (HRCT) images in the axial plane (A and B) reveal inconclusive posterior basal subpleural honeycombing images. Additional HRCT acquisition in the prone position (C and D) confirms the presence of honeycombing (arrows).
Traction bronchiolectasis/bronchiectasis: represent irregular dilatation of bronchi and bronchioles caused by adjacent retractile fibrosis.14 They are a key finding in the diagnosis of pulmonary fibrosis and the pattern of probable UIP, as well as an important prognostic marker in the UIP pattern.18 In the UIP pattern, they are typically peripherally located, which differentiates them from NSIP, where they tend to have a more central distribution. In UIP they are varicose in appearance and coexist with honeycombing, reticulation, and ground-glass opacities. Differentiating them from honeycombing often requires the help of multiplanar reconstructions and additional reconstructions, such as the minimum intensity projection, which facilitate the identification of the communication with the bronchial structures.16
Reticular opacities: are characterised by a network of fine lines due to irregular thickening of the interlobular and intralobular septa.4
Ground-glass opacities: although ground glass is not characteristic of the UIP pattern, it can coexist with traction bronchiectasis, reticulation, and honeycombing, forming part of the fibrosis process.19 When the ground-glass pattern is the predominant finding or is present in areas without fibrotic involvement, an acute exacerbation (AE) or infection should be suspected.3,4,20
Other findings present in IPFCalcifications: small foci of nodular calcifications can be seen in areas of fibrosis as a result of pulmonary ossification (dendriform pulmonary ossification),21 the prevalence of these calcifications being significantly higher in patients with UIP (28.5%) than in those with other fibrosing DILD (8.3%, P<.001).22 This finding was recently included in the UIP pattern of the 2022 ATS guidelines.
Mediastinal adenopathies: enlarged mediastinal lymph nodes are common in DILD and are present in up to 70% of patients with a UIP pattern.23 A higher prevalence of lymphadenopathy has been described in the group of patients with IPF when compared to the group with other DILDs. Likewise, it has been demonstrated that patients with DILD and mediastinal nodes larger than 10mm had higher mortality, worse lung function, and a higher risk of hospitalisation than patients without enlarged nodes.24
UIP patterns on HRCTBy evaluating the HRCT findings, one of the four categories of the UIP pattern included in the IPF diagnostic guidelines must be assigned. The main guidelines for the radiologist are those of the Fleischner Society4 and the 2018 and 2022 ATS/ERS/JRS/ALAT guidelines3,7 (Table 1).
Comparison of the UIP pattern categories on HRCT of the 2018 Fleischner Society consensus guidelines and the 2022 ATS/ERS/JRS/ALAT guidelines.
| HRCT pattern | White paper Fleischner Society 2018 | ATS/ERS/JRS/ALAT 2022 Guidelines | ||
|---|---|---|---|---|
| Distribution | Characteristics | Distribution | Characteristics | |
| UIP or typical of UIP | Basal and subpleural predominance, often heterogeneous, may be diffuse | Honeycombing. Reticulation, traction bronchiolectasis or bronchiectasis | Basal and subpleural predominance, often heterogeneous | Honeycombing with or without traction bronchiolectasis/bronchiectasis |
| Absence of findings indicative of another diagnosis | May be diffuse and asymmetric | Irregular thickening of interlobular septa | ||
| Usually with overlapping reticular pattern, mild ground-glass opacities | ||||
| May have pulmonary ossification | ||||
| Probable UIP | Basal and subpleural predominance, often heterogeneous | Reticulation, traction bronchiolectasis or bronchiectasis | Basal and subpleural predominance, often heterogeneous | Reticulation with traction bronchiolectasis/bronchiectasis |
| Absence of honeycombing | May have mild ground-glass opacities | |||
| Absence of findings indicative of another diagnosis | Subpleural areas are not preserved | |||
| Indeterminate for UIP | Variable or diffuse (without subpleural or basal predominance) | Reticulation with some discrete findings indicating a non-UIP pattern | Diffuse distribution without subpleural predominance | Characteristics of pulmonary fibrosis that do not indicate a specific aetiology |
| Inconsistent with diagnosis of IPF or indicative of alternative diagnosis | Upper-middle lung predominance | Predominant consolidation | Peribronchovascular predominance with subpleural preservation (NSIP) | Cysts (LAM, PLCH, LIP, DIP) |
| Peribronchovascular predominance with subpleural preservation | Extensive ground-glass opacities | Perilymphatic distribution (sarcoidosis) | Mosaic attenuation or three-density pattern (HP) | |
| Extensive mosaic attenuation with air trapping on expiration | Upper or middle lung (fibrotic HP, collagen diseases, sarcoidosis) | Predominant ground-glass (HP, smoking-related ILD, drug toxicity, acute exacerbation) | ||
| Diffuse nodules or cysts | Subpleural preservation (NSIP or smoking-related ILD) | Centrilobular micronodules (HP or smoking-related ILD) | ||
| Nodules (sarcoidosis) | ||||
| Consolidation (OP) | ||||
| Pleural plaques | ||||
| Oesophageal dilatation | ||||
DIP: desquamative interstitial pneumonia; HP: hypersensitivity pneumonitis; HPCL: pulmonary Langerhans cell histiocytosis; ILD: interstitial lung disease; LAM: lymphangioleiomyomatosis; LIP: lymphocytic interstitial pneumonia; NSIP: non-specific interstitial pneumonia; OP: organising pneumonia; UIP: usual interstitial pneumonia.
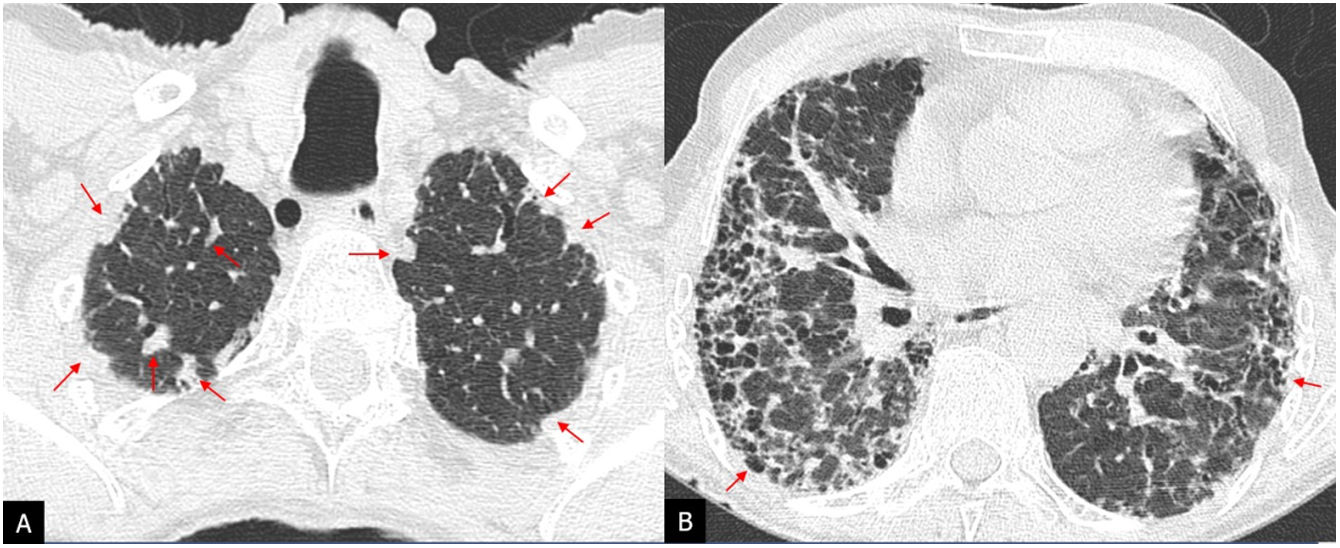
UIP or typical UIP pattern: reticular opacities and honeycomb cysts, with traction bronchiectasis and peripheral subpleural distribution, predominantly basal; there should be no other findings indicative of an alternative diagnosis (Fig. 3).
 of three patients diagnosed with idiopathic pulmonary fibrosis (A–C). HRCT in the axial plane with basal honeycomb cysts (arrows), being more prominent in patient A and scanty in patient C.")
Pattern of probable UIP: includes the same findings as the UIP pattern, except for honeycombing. The precise HRCT findings that this pattern should present have been described: predominantly basal reticulation, heterogeneous distribution (patchy, alternating with unaffected areas, related to the temporal and spatial heterogeneity of the histological UIP pattern) and non-segmental (without fissures preserved), lobular distortion and presence of traction bronchioloectasis and bronchiectasis (Fig. 4).
 High-resolution computed tomography (HRCT) in the axial plane revealing traction bronchiolectasis with predominantly basal subpleural reticular opacities (arrows). Honeycombing not identified. B) HRCT in the axial plane with a pattern of probable UIP with peripheral calcifications indicative of dendriform pulmonary ossification (arrows). C and D) Pattern of probable UIP in HRCT and minimal intensity projection (minIP) reconstruction that better reveals traction bronchiolectasis and bronchiectasis (arrows).")
Pattern of probable usual interstitial pneumonia in three patients with idiopathic pulmonary fibrosis. A) High-resolution computed tomography (HRCT) in the axial plane revealing traction bronchiolectasis with predominantly basal subpleural reticular opacities (arrows). Honeycombing not identified. B) HRCT in the axial plane with a pattern of probable UIP with peripheral calcifications indicative of dendriform pulmonary ossification (arrows). C and D) Pattern of probable UIP in HRCT and minimal intensity projection (minIP) reconstruction that better reveals traction bronchiolectasis and bronchiectasis (arrows).
Indeterminate pattern for UIP: appears when the fibrosis does not have a predominantly peripheral subpleural or basal distribution or when it coexists with other findings, such as consolidations, extensive ground-glass opacities, nodules, non-honeycomb cysts, or areas of air trapping. This pattern presents characteristics that suggest its differential diagnosis with other fibrosing diseases, mainly fibrotic hypersensitivity pneumonitis and NSIP. Early UIP is included in this pattern in the ATS/ERS/JRS/ALAT guidelines from 20183 (Fig. 5).
 Axial high-resolution computed tomography (HRCT) reveals extensive ground-glass and peribronchovascular reticular opacities associated with traction bronchiectasis with less posterior basal involvement (arrows). C and D) HRCT in the axial plane with faint subpleural reticular opacities of basal predominance (arrows) without traction bronchiectasis.")
Indeterminate pattern of usual interstitial pneumonia in two patients with a diagnosis of idiopathic pulmonary fibrosis diagnosed by lung biopsy. A and B) Axial high-resolution computed tomography (HRCT) reveals extensive ground-glass and peribronchovascular reticular opacities associated with traction bronchiectasis with less posterior basal involvement (arrows). C and D) HRCT in the axial plane with faint subpleural reticular opacities of basal predominance (arrows) without traction bronchiectasis.
Alternative diagnostic pattern or pattern with characteristics inconsistent with the diagnosis of IPF: in these cases, the radiological findings and their distribution point towards another type of fibrosis. For example, fibrosis with a peribronchovascular distribution predominantly in the upper lung fields, with ground-glass opacities (in the absence of exacerbation) associated with air trapping in non-fibrotic areas, suggests fibrotic hypersensitivity pneumonitis (Fig. 6).
 Ground-glass opacities of peripheral distribution in a patient with eosinophilic pneumonia (arrows). B) Ground-glass and peripheral reticular opacities and bibasal traction bronchiolectasis with the subpleural zone preserved (arrows) in a patient with a pattern of non-specific interstitial pneumonia. C) Traction bronchiectasis with pronounced peribronchovascular parenchymal distortion in the upper lung fields (arrows) in a patient with fibrotic sarcoidosis. D) Peribronchovascular ground-glass patchy foci (arrows) in a patient with organising pneumonia.")
Pattern indicative of alternative diagnosis to idiopathic pulmonary fibrosis. High-resolution computed tomography images in the axial plane of four patients. A) Ground-glass opacities of peripheral distribution in a patient with eosinophilic pneumonia (arrows). B) Ground-glass and peripheral reticular opacities and bibasal traction bronchiolectasis with the subpleural zone preserved (arrows) in a patient with a pattern of non-specific interstitial pneumonia. C) Traction bronchiectasis with pronounced peribronchovascular parenchymal distortion in the upper lung fields (arrows) in a patient with fibrotic sarcoidosis. D) Peribronchovascular ground-glass patchy foci (arrows) in a patient with organising pneumonia.
Each of these patterns has a positive predictive value (PPV) for histological UIP.7 It is greater than 90% in the UIP pattern, and if there is no known cause of fibrosis, it allows the diagnosis of IPF without needing a lung biopsy. The PPV in the pattern of probable UIP is approximately 80%. In an adequate clinical context, and without known causes of fibrosis, it enables the diagnosis of UIP without needing a lung biopsy. The indeterminate pattern for UIP has a PPV of approximately 50%, and the alternative diagnostic design less than 50%. Therefore, to diagnose IPF in these two groups of patients, it will be necessary to perform a lung biopsy. The final diagnosis will depend on the combination of radiopathological findings.7
The latest update to the ATS/ERS/JRS/ALAT guidelines, in May 2022,7 includes minor changes in the radiological patterns of UIP compared to those of 2018.3Table 1 shows the main characteristics of the HRCT patterns and the differences between the Fleischner4 and ATS/ERS/JRS/ALAT guidelines from 2022.
In any radiological report evaluating possible IPF, the UIP pattern should be included, as well as the possible differential diagnosis if the pattern is indeterminate or not indicative of IPF.
Differential diagnosis of the UIP pattern and IPFAlthough the UIP pattern is characteristic, it is not exclusive to IPF. We can see it in other interstitial lung diseases, mainly in fibrotic NSIP, fibrotic hypersensitivity pneumonitis (FHP), collagen diseases (especially rheumatoid arthritis) and interstitial pneumonia with autoimmune features (IPAF), asbestosis, stage IV sarcoidosis, and drug-related and exposure diseases.25 The most important and common differential diagnosis is with fibrotic NSIP, with FHP, and with collagen diseases. The recognition of additional findings other than the UIP pattern in the HRCT can guide us towards an alternative diagnosis to IPF25,26 (Table 2).
Additional HRCT findings associated with the UIP pattern that indicate an alternative diagnosis to IPF.
| Additional findings | Probable diagnosis |
|---|---|
| Centrilobular nodules | Fibrotic hypersensitivity pneumonitis |
| Mosaic attenuation-air trapping pattern (especially in areas without fibrosis) | |
| Fibrosis with relatively preserved lung bases | |
| Calcified pleural plaques | Asbestosis |
| Exuberant honeycombing | ILD associated with collagen diseases |
| Atypical distribution of the UIP pattern: | |
| “Anterior upper lobe sign” | |
| “Straight edge” sign | |
| Pleural effusion. Pleural thickening | |
| Oesophageal dilatation | |
| Calcified adenopathies | Silicosis-sarcoidosis |
| Pulmonary nodules of perilymphatic distribution | |
| Emphysema with/without ground-glass | Smoking-related ILD. CPFE |
| Apical pleuroparenchymal thickening | PPFE |
| Distal clavicular erosion | RA-related ILD |
| Hyperattenuation of the liver parenchyma | Amiodarone toxicity |
CPFE: combination pulmonary fibrosis and emphysema; HRCT: high-resolution computed tomography; ILD: interstitial lung disease; IPF: idiopathic pulmonary fibrosis; PPFE: pleuroparenchymal fibroelastosis; RA: rheumatoid arthritis; UIP: usual interstitial pneumonia.
Honeycombing may be present in NSIP, although it is mild and not the predominant finding, as are ground-glass opacities associated with traction bronchiectasis. Although the findings coincide with those of IPF in that the basal location is the most common, ground-glass opacities tend to leave the subpleural space preserved, and traction bronchiectasis have a patchy distribution and are more peribronchovascular than subpleural, as in the case of UIP.5 When NSIP presents fibrotic progression, the fibrosis findings adopt an arrangement similar to that of the UIP pattern, so it is important to have access to previous examinations to assess the evolution.27
Interstitial lung disease associated with collagen diseasesCollagen diseases and IPAF will manifest more commonly with a pattern of NSIP or organising pneumonia, or a combination of both. In the event that they present with a UIP pattern, three radiological signs have been described that are more frequent in these entities than in IPF. These are the exuberant honeycombing sign, the straight edge sign, and the anterior upper lobe sign (Fig. 7). Exuberant honeycombing refers to pulmonary fibrosis almost exclusively in the form of honeycombing, constituting extensive honeycombing that affects more than 70% of the fibrotic areas of the lung; the anterior upper lobe sign is fibrosis predominantly in the anterior regions of the upper lobes with concomitant involvement of the lower lobes, and the straight edge sign is the delineation of a sharp border separating areas of fibrosis from healthy lung in the craniocaudal plane.28
 in the axial plane showing extensive honeycombing (arrows) with a basal predominance (A) and HRCT image in the coronal plane with sharp delineation of the honeycombing from healthy lung parenchyma (arrows).")
Exuberant honeycombing sign and straight edge sign in a patient with mixed connective tissue disease. High-resolution computed tomography (HRCT) in the axial plane showing extensive honeycombing (arrows) with a basal predominance (A) and HRCT image in the coronal plane with sharp delineation of the honeycombing from healthy lung parenchyma (arrows).
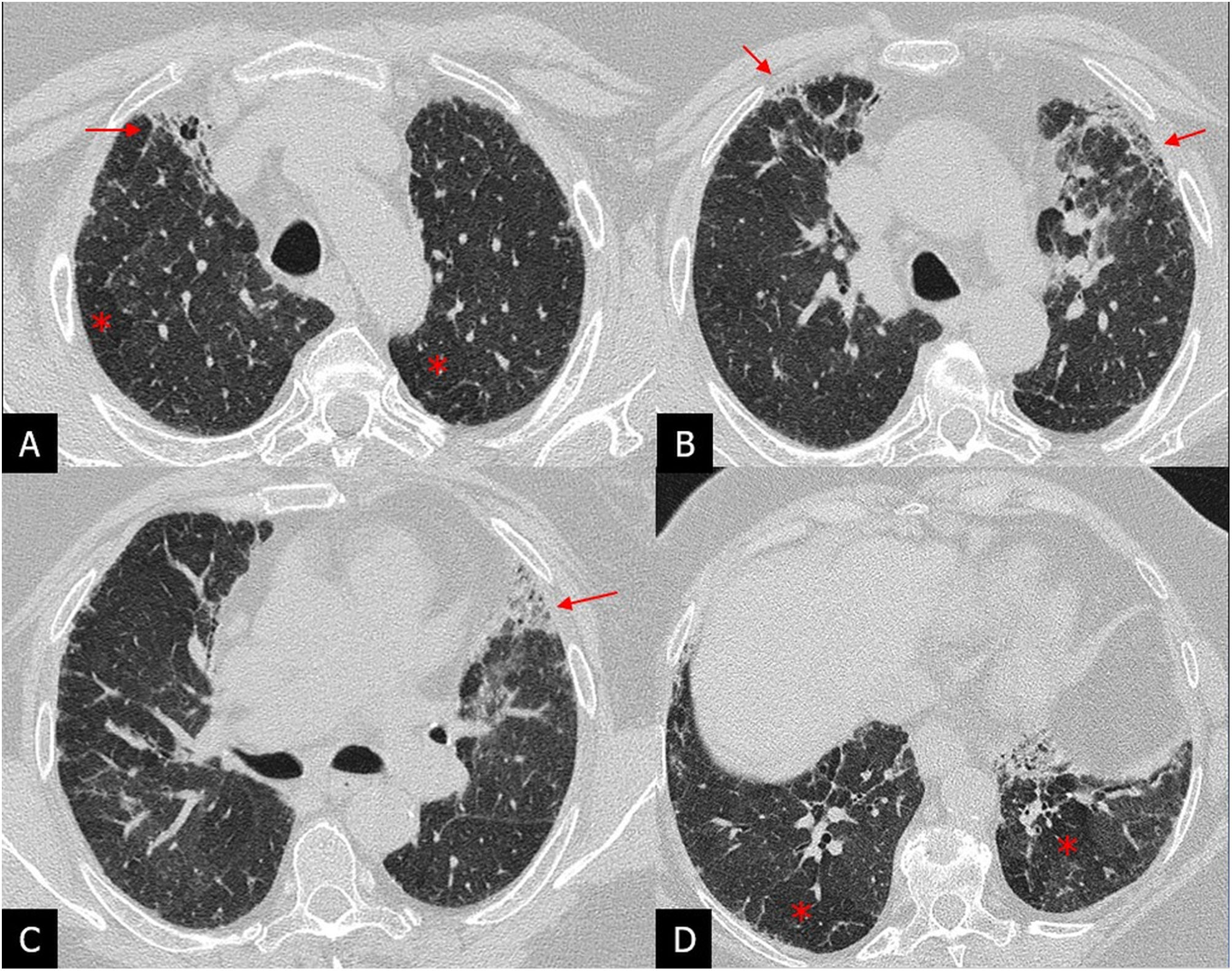
Although classically, it has been considered that fibrosis in FHP predominates in the upper lung fields, being a distinctive feature of IPF,29 FHP-associated fibrosis has been found to affect the lower lung fields in more than 50% of cases. Honeycombing may be present and extensive in severe forms and indistinguishable from that of IPF.30 The UIP pattern has been included in the 2020 ATS/JRS/ALAT multidisciplinary guidelines for the diagnosis of HP in the category of HRCT compatible with FHP.31 Additional findings that should lead to suspicion of FHP in a UIP pattern are the presence of small airway involvement in the form of poorly-defined centrilobular nodules or the mosaic attenuation pattern of lobular morphology or the three-density pattern, highly specific for NHF, or air trapping on expiration, especially if present in areas without fibrosis31,32 (Fig. 8).
 High-resolution computed tomography axial plane images revealing signs of patchy fibrosis with traction bronchiolectasis and small honeycomb cysts (arrows) and a mosaic attenuation pattern with areas of hypoattenuation (asterisks) indicative of air trapping.")
Patient with fibrotic hypersensitivity pneumonitis diagnosed by lung biopsy. A–D) High-resolution computed tomography axial plane images revealing signs of patchy fibrosis with traction bronchiolectasis and small honeycomb cysts (arrows) and a mosaic attenuation pattern with areas of hypoattenuation (asterisks) indicative of air trapping.
Sometimes a lung biopsy will be required to make the diagnosis of IPF when the clinical history and radiology are not sufficient.
Obtaining lung parenchyma for diagnosis in DILDs can be performed by transbronchial biopsy with conventional forceps or with the cryobiopsy technique, as well as by video-assisted surgery. The information from the imaging and histology are the basis for the multidisciplinary committee to reach a diagnosis in all patients with suspected IPF.
Since 2011, in the same way, that occurs in the field of radiology, the diagnosis is made with degrees of probability, in relation to the presence of all the characteristics of the UIP or only some of them, following the criteria defined in the literature that were agreed upon in 2018.3 The concepts for the pathologist to define a certain biopsy as a pattern of UIP, probable UIP, indeterminate for UIP and alternative diagnoses are indicated in Table 3. A biopsy can have all the UIP criteria, but if it also shows some contradictory histological data, such as granulomas or hyaline membranes, it is no longer a pattern of UIP.
Classification of UIP pathological findings.
| UIP pattern | Probable UIP | Indeterminate for UIP | Alternative diagnoses |
|---|---|---|---|
| Dense fibrosis with architectural distortion, predominantly subpleural or paraseptal | Any of the characteristics from column 1 | Fibrosis with or without architectural distortion with data indicating non-UIP disease or secondary UIPa | Characteristics of other types of unusual interstitial pneumonia |
| Heterogeneous fibrotic involvement | In addition to: absence of data indicating an alternative diagnosis | Presence of any data from column 1, along with data indicating an alternative diagnosisb | Histological data of other diseases: HP, LAM, PLCH |
| Presence of fibroblastic foci | Or only: honeycombing | ||
| Absence of data indicating an alternative diagnosis |
HP: hypersensitivity pneumonitis; LAM: lymphangioleiomyomatosis; PLCH: pulmonary Langerhans cell histiocytosis; UIP: usual interstitial pneumonia.
Modified from Raghu et al.3
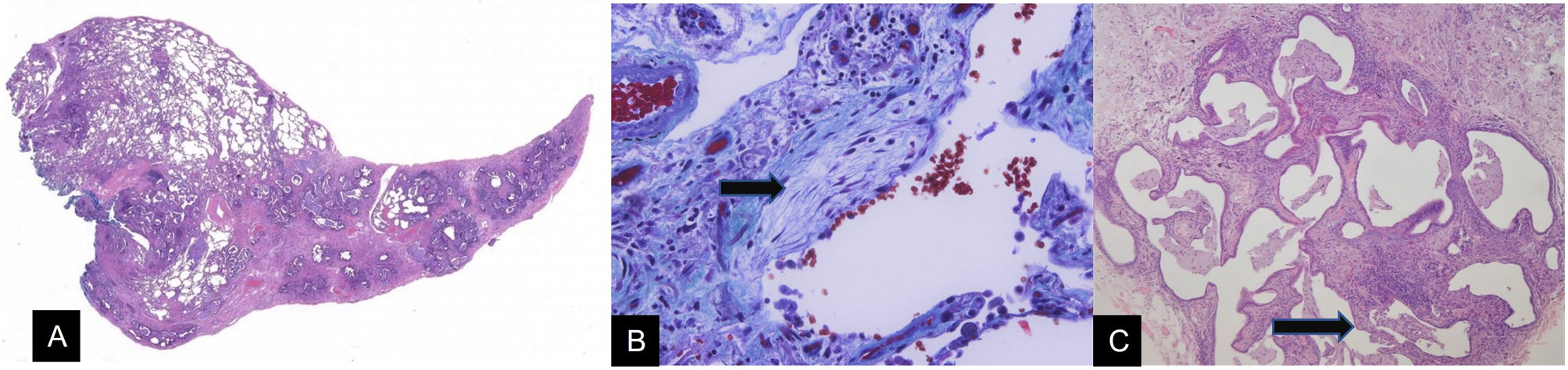
The UIP pattern is based on the heterogeneous distribution of lesions in the lung parenchyma so that lesions with a greater or lesser degree of involvement are simultaneously seen, even mixed with areas of healthy parenchyma. These heterogeneous lesions are made up of fibrosis in the interstitium, which causes distortion of the lung architecture. There is always a microscopic tendency towards predominantly subpleural or paraseptal involvement, which is more serious than in the central areas of the secondary lobule (Fig. 9A). Fibrous tissue has a clear tendency to deposit collagen fibres. However, it is also associated with foci of cell proliferation with an amorphous matrix and without collagen fibres, which are called fibroblastic foci (Fig. 9B). In the latest classifications these foci have become a fundamental point for the diagnosis of the UIP pattern. The maximum alteration of the lung parenchyma consists of the development of areas of the so-called honeycombing, a term that reflects a situation of large, pseudocystic air spaces with rigid walls (Fig. 9C). This honeycombing corresponds to the final stage of architectural alteration seen in interstitial pneumonias. Histologically, the abnormal air spaces are large and lined with bronchiolar epithelium. Mucoid secretory material from the epithelial lining cells progressively accumulates in these air spaces. The resulting image is of variable occupation of the spaces by retained mucoid material, with a mixture of free, ventilated air spaces and spaces occupied by accumulated secretions. The last component is the mononuclear inflammatory infiltrate of irregular distribution without forming significant aggregates. This last aspect is relevant since the presence of lymphocyte aggregates with germinal centres opens up various options for differential diagnosis, among which the association of DILD with systemic autoimmune diseases, such as rheumatoid arthritis, stands out.
 Histological image at low magnification of a usual interstitial pneumonia (UIP) pattern. Predominantly subpleural architectural distortion can be seen, with heterogeneous involvement of the lung parenchyma (H&E 10×). B) High magnification image of an area of lung parenchyma with inflammation and fibrosis, revealing a fibroblastic focus (arrow), which is identified by the paleness of its matrix. Mallory Trichrome 400×. C) Close-up image of an area of pulmonary honeycombing (arrow) corresponding to a UIP pattern. In this lung, the heterogeneity of the inflammatory and fibrosing component stands out, with focal preservation of some septa (H&E 40×).")
A) Histological image at low magnification of a usual interstitial pneumonia (UIP) pattern. Predominantly subpleural architectural distortion can be seen, with heterogeneous involvement of the lung parenchyma (H&E 10×). B) High magnification image of an area of lung parenchyma with inflammation and fibrosis, revealing a fibroblastic focus (arrow), which is identified by the paleness of its matrix. Mallory Trichrome 400×. C) Close-up image of an area of pulmonary honeycombing (arrow) corresponding to a UIP pattern. In this lung, the heterogeneity of the inflammatory and fibrosing component stands out, with focal preservation of some septa (H&E 40×).
Multidisciplinary assessment of a patient with fibrosing interstitial lung disease is essential to establish the diagnosis and determine the need for a biopsy or other diagnostic tests. To assess HRCT patterns and the need for biopsy, it is essential to include the appropriate clinical context of IPF, the absence of environmental or drug exposure, and the exclusion of collagen diseases.3,4
The Fleischner guidelines introduce the concept of a working diagnosis or provisional diagnosis of IPF in those cases where a biopsy cannot be obtained in fibrosing interstitial lung disease without a characteristic pattern on HRCT and in the absence of an alternative diagnosis. This working diagnosis should be reviewed at regular intervals, to confirm IPF or change the diagnosis to another fibrosing lung disease.
The 2018 ATS/ERS/JRS/ALAT guidelines define specific combinations of HRCT patterns and histopathological patterns in patients with lung biopsy. In addition, the latest guide for 2022 takes into account the importance of transbronchial cryobiopsy, it being less invasive and expensive than surgical biopsy, including it in the diagnostic algorithm as an alternative to surgical biopsy in centres with experience in the procedure and interpretation of the samples,7 coinciding with what was previously published in Spain.33
Pathological entities associated with IPFPleuroparenchymal fibroelastosisPatients with a UIP pattern may have associated features of pleuroparenchymal fibroelastosis (PPFE) with typical pleuroparenchymal thickening predominantly in the upper lobes, dense subpleural consolidations with traction bronchiectasis, and loss of volume. PPFE is associated with IPF in 6%–10% of cases, and poorer lung function and a worse prognosis have been described in these patients compared to patients without PPFE.34 Patients who meet IPF criteria should be considered as IPF patients even if they have an associated UIP and PPFE pattern since this overlap has not yet been considered an independent entity3,4 (Fig. 10).
 in a patient with idiopathic pulmonary fibrosis. High-resolution computed tomography in the axial plane revealing apical pleuroparenchymal thickening in A (arrows) with a predominantly basal UIP pattern with honeycomb cysts in B (arrows).")
Pleuroparenchymal fibroelastosis associated with a pattern of usual interstitial pneumonia (UIP) in a patient with idiopathic pulmonary fibrosis. High-resolution computed tomography in the axial plane revealing apical pleuroparenchymal thickening in A (arrows) with a predominantly basal UIP pattern with honeycomb cysts in B (arrows).
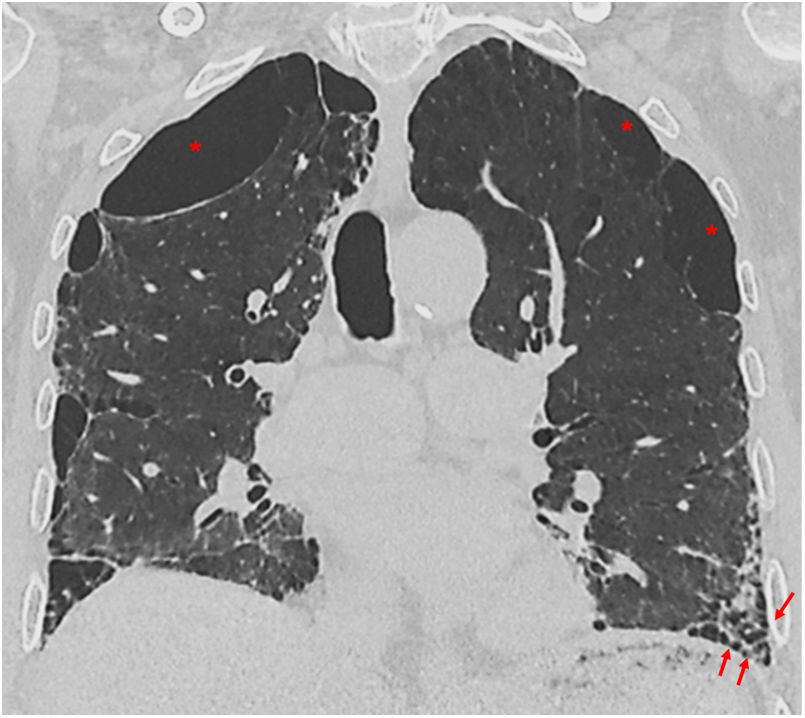
Many patients with IPF are current or former smokers and may present with smoking-related lung disease in association with fibrosis. Approximately a third of patients with IPF have emphysema.35 The entity of combined pulmonary fibrosis and emphysema (CPFE) classically corresponds to the coexistence of emphysema in the upper lobes with a predominantly basal UIP pattern (Fig. 11). The concept of CPFE has recently been reviewed in a multidisciplinary document published by the ATS/ERS/JRS/ALAT and proposes expanding the entity to different patterns of pulmonary fibrosis (reticulation or ground-glass, honeycombing or traction bronchiectasis) defined by HRCT or pathology (fibrotic NSIP, fibrotic HP, etc.).36 Patients with CPFE tend to have a different clinical and radiological course compared to those with emphysema or IPF alone, with a higher incidence of comorbidities such as pulmonary hypertension (PHTN) and lung cancer, so it is important to recognise this entity on HRCT, as it can change the patient's prognosis and treatment.36
HRCT follow-up. Complications of IPF.High-resolution computed tomography in the coronal plane with severe paraseptal emphysema, subpleural bullae (asterisks), and usual interstitial pneumonia pattern at the lung bases (arrows).")
The use of HRCT in clinical practice for the routine follow-up of patients with IPF is controversial and not recommended in stable patients. Radiological progression is a negative prognostic factor for survival. Annual follow-up can serve as a prognostic marker and to identify complications such as lung cancer. HRCT is indicated in the follow-up of these patients when they present with unexpected clinical/functional deterioration, to assess complications such as AE, when there is a non-definitive radiopathological or radiological diagnosis of UIP, a provisional diagnosis, and when evolution is awaited to confirm the diagnosis.4,37
Acute exacerbationThe current definition of AE is clinically significant respiratory deterioration, which is acute, typically in less than a month, and which is characterised by the appearance of new alveolar opacities.38 Histopathologically, the characteristic pattern is diffuse alveolar damage. It is a clinical picture that is not exclusive to IPF and can occur in any chronic interstitial lung disease. On HRCT we will see ground-glass opacities or bilateral consolidations on an underlying fibrotic lung. The clinical and radiological picture must be correctly assessed so as not to report the CT as “more suggestive of a non-IPF diagnosis or an alternative diagnosis” given the presence of ground-glass opacities and consolidation, and to assess it as AE of previous fibrosing interstitial lung disease.5 In the absence of a previous HRCT, bilateral ground-glass opacities or bilateral consolidations in a UIP pattern are highly indicative of AD (Fig. 12).
 High resolution computed tomography in the axial plane acquired prior to the episode of acute exacerbation revealing a pattern of usual interstitial pneumonia with subpleural honeycomb cysts (arrows). C and D) CT pulmonary angiogram to rule out pulmonary thromboembolism with the appearance of extensive ground-glass opacities superimposed on the interstitial pattern indicative of an acute exacerbation.")
Acute exacerbation in a patient with idiopathic pulmonary fibrosis undergoing antifibrotic treatment. A and B) High resolution computed tomography in the axial plane acquired prior to the episode of acute exacerbation revealing a pattern of usual interstitial pneumonia with subpleural honeycomb cysts (arrows). C and D) CT pulmonary angiogram to rule out pulmonary thromboembolism with the appearance of extensive ground-glass opacities superimposed on the interstitial pattern indicative of an acute exacerbation.
IPF is associated with a higher incidence of lung cancer, especially in current and former smokers. Lung cancer occurs in around 10% of patients with IPF and significantly worsens the prognosis of the disease.39 Squamous cell carcinoma seems to be more common and usually involves peripheral lesions predominantly in the lower lobes, in areas of fibrosis, unlike what occurs in the general population, in which it predominates in the upper lobes.40 Since cancers in IPF have a propensity to arise in areas adjacent to fibrosis or in regions of fibrosis with distortion of lung architecture, delay in diagnosis is common.41 Given the suspicion of cancer in a patient with IPF, it is advisable to recommend early follow-up CT or positron emission tomography-CT.
Other complications in these patients are mycobacterial infections or infections due to Aspergillus, pneumothorax or pneumomediastinum, pulmonary thromboembolism (PTE) and PHTN. Spontaneous pneumothorax and pneumomediastinum occurring in these patients are considered poor prognostic events.42,43 Although PHTN in IPF is not usually serious, the demonstration of a ratio between the diameter of the pulmonary artery and that of the aorta greater than 1 (PA:Ao >1) is associated with a worse evolution in patients with IPF.44
Since PTE and AE should be ruled out in the event of acute worsening of dyspnoea in a patient with known IPF, it is recommended that a CT pulmonary angiogram be acquired, alone or in combination with HRCT, for evaluation of the lung parenchyma.3
ConclusionsThe radiologist plays a fundamental role in diagnosing IPF. HRCT is the fundamental tool and we need to understand the different radiological patterns of IPF in its most current classification and its differential diagnosis with other fibrosing interstitial lung diseases. The typical and probable UIP patterns in the appropriate clinical setting obviate the need for surgical biopsy. Patterns that are indeterminate and not indicative of IPF, and any radiological pattern outside the clinical context of IPF, will require multidisciplinary discussion and lung biopsy assessment for diagnosis. Early diagnosis is important for the establishment of the necessary treatment. Follow-up CT is not clearly indicated, although it may offer prognostic information or be necessary to reach a diagnosis based on evolution in patients who are not eligible for biopsy.
AuthorshipAll authors have made substantial contributions in the following aspects: 1) study conception and design, or data acquisition, or data analysis and interpretation; 2) the drafting of the article or the critical revision of the intellectual content, AND 3) final approval of the version submitted.
Conflicts of interestThe authors declare that they have no conflicts of interest.

