




To review the imaging findings for the different types of pulmonary histiocytosis. In particular, in addition to the well-known pulmonary Langerhans cell histiocytosis related to smoking and its possible appearance in nonsmokers, we focus on non-Langerhans cell histiocytosis in Rosai–Dorfman disease and Erdheim–Chester disease. We also review the etiopathogenesis, histology, clinical presentation, and treatment of pulmonary histiocytosis.
ConclusionLangerhans cell histiocytosis, Rosai–Dorfman disease, and Erdheim–Chester disease are idiopathic diseases in which the proliferation and infiltration of histiocytes is the histologic finding that confirms the diagnosis. Langerhans cell histiocytosis manifests as nodules and cysts that spare the costophrenic angles; it typically appears in smokers. Although it is uncommon in nonsmokers, Langerhans cell histiocytosis should also be considered in nonsmokers treated with chemotherapy and radiotherapy in whom cavitated nodules appear and should be included in the differential diagnosis together with metastatic disease and opportunistic infections. Rosai–Dorfman disease and Erdheim–Chester disease present with less specific thoracic findings such as adenopathies, interstitial thickening, and pleural effusion. In Erdheim–Chester disease, the characteristic extrathoracic manifestations are usually key for the diagnosis.
Revisar los hallazgos de imagen de las diferentes histiocitosis pulmonares. En concreto, además de la conocida histiocitosis de células de Langerhans relacionada con el tabaco y su posible aparición sin antecedentes de este, la enfermedad de Rosai-Dorfman y la enfermedad de Erdheim-Chester. También se revisa su etiopatogenia, histología, clínica y tratamiento.
ConclusiónLa histiocitosis de células de Langerhans, la enfermedad de Rosai-Dorfman y la enfermedad de Erdheim-Chester son un conjunto de patologías de causa idiopática en las que la proliferación e infiltración de histiocitos es el hallazgo anatomopatológico diagnóstico. La histiocitosis de células de Langerhans se manifiesta en forma de nódulos y quistes que respetan los ángulos costofrénicos, característicamente en pacientes fumadores. Aunque es poco frecuente, debe pensarse en esta entidad en pacientes no fumadores, en tratamiento quimio y radioterapéutico, con nódulos cavitados de nueva aparición e incluirse en el diagnóstico diferencial junto con la enfermedad metastásica y la infección oportunista. La enfermedad de Rosai-Dorfman y la enfermedad de Erdheim-Chester se presentan con hallazgos torácicos más inespecíficos, como adenopatías, engrosamiento intersticial y derrame pleural. En la enfermedad de Erdheim-Chester, las características manifestaciones extratorácicas suelen ser claves en el diagnóstico.
Histiocytosis is a group of diseases characterised by abnormal proliferation of histiocytes with infiltration of a specific organ. This group of disorders derives from the phagocytic mononuclear system, the main roles of which are phagocytosis of foreign material, antigen processing and antigen presentation to lymphocytes. The mononuclear phagocyte system includes macrophages and dendritic cells. Macrophages are distributed all around the body, but they are found mainly in mucous membranes and other potential gateways for microorganisms, where they function as innate and specific immunity. In the lungs, they are usually present in the alveoli. Dendritic cells are located primarily in the skin, mucous membranes, bone marrow, spleen, thymus gland and lymph nodes, and, although they have a less important role in phagocytosis, they are more important at the beginning of the T cell-dependent response.1 Langerhans cells (LC) are specifically derived from dendritic cells and are found in the epidermal layer of the skin. Langerhans cell histiocytosis (LCH), which is typically associated with smoking and manifests itself with distinctive radiological findings, is the most common and well-known condition in which these cells are involved. An association has been reported between LCH and certain types of cancer, such as Hodgkin's lymphoma,2–5 and after treatment with chemotherapy and radiotherapy.6,7 LCH is not the only histiocytosis which affects the respiratory system. Other lesser-known types of pulmonary histiocytosis are Rosai–Dorfman disease (RDD) (derived from macrophages) and Erdheim–Chester disease (ECD) (derived from non-Langerhans dendritic cells). The secondary and malignant forms of histiocytosis are not discussed in this manuscript; we have focused on primary non-neoplastic pulmonary histiocytosis in adults (Table 1).8,9
Summary of primary histiocytic disorders of non-neoplastic (or unknown) aetiology and those with malignant behaviour.
| Primary histiocytic disorders |
| Non-neoplastic or unknown aetiology |
| Langerhans cell histiocytosis |
| Adult form (lung involvement)a |
| Paediatric form (multisystem involvement) |
| Letterer–Siwe disease |
| Hand–Schüller–Christian disease |
| Hashimoto–Pritzker syndrome |
| Rosai–Dorfman diseasea |
| Erdheim–Chester diseasea |
| Histiocytic neoplasms |
| Follicular dendritic cell tumour |
| Histiocytic sarcoma |
| Langerhans cell sarcoma |
| Reticulum cell sarcoma |
The aim of our study was to review the imaging findings for the different thoracic manifestations of the most common types of histiocytosis, and provide the keys to an accurate diagnosis. We also describe the aetiopathogenesis, histology, clinical presentation and treatment.
Langerhans cell histiocytosisLCH is the most common disorder associated with dendritic cells.10 Cell behaviour in LCH varies according to the age of the patient. In adults, the pathological LC behave reactively, with no neoplastic growth, and the most commonly, and usually the only, affected organ is the lung. In contrast, in childhood, proliferation of pathological LC is characterised by their clonal neoplasm behaviour8 and multisystem involvement. Letterer–Siwe and Hand–Schüller–Christian diseases and Hashimoto–Pritzker syndrome are LCH with multisystem involvement which typically begin in childhood. Lung involvement is rare in these cases and, if it does occur, is part of the systemic condition and unrelated to smoking.
In this article, we focus on LCH with lung involvement in adults.
AetiopathogenesisAlthough the aetiology is unknown, a possible antigenic cause, somatic mutations or infectious aetiology have all been suggested.11,12 LCH occurs almost exclusively in smokers or former smokers, which supports the antigenic hypothesis. The influence of smoking on the development and progression of the disease is reinforced by the fact that it progresses more rapidly in patients who continue to smoke and regresses in the majority of those who stop.2,3,6,7,12
Much less common and understood is the development of LCH in non-smoking patients.3 Its association with different types of cancer5 and lymphoproliferative processes, specifically Hodgkin's lymphoma, has been described,2,4,6,13,14 particularly after chemotherapy and radiotherapy.6,7
HistologyIn early stages, LCH is characterised by the proliferation and infiltration of the respiratory bronchiole and the adjacent interstitium by LC, eosinophils and macrophages, while in the later stages fibrosis predominates.
The LC have differentiating microscopic and biochemical characteristics: a convoluted nuclear morphology, with the characteristic image of wrinkled paper (Figs. 1 and 2).8 They also have Birbeck granules and quintuple-layered intracytoplasmic inclusion bodies which can be seen by optical microscopy.8 The “tennis racket” image corresponds in electronic microscopy with the fusion of the Birbeck granules with intracytoplasmic vesicles.3,10 The immunohistochemical study is positive for S100, CD1a and langerin antigen (Fig. 2).8
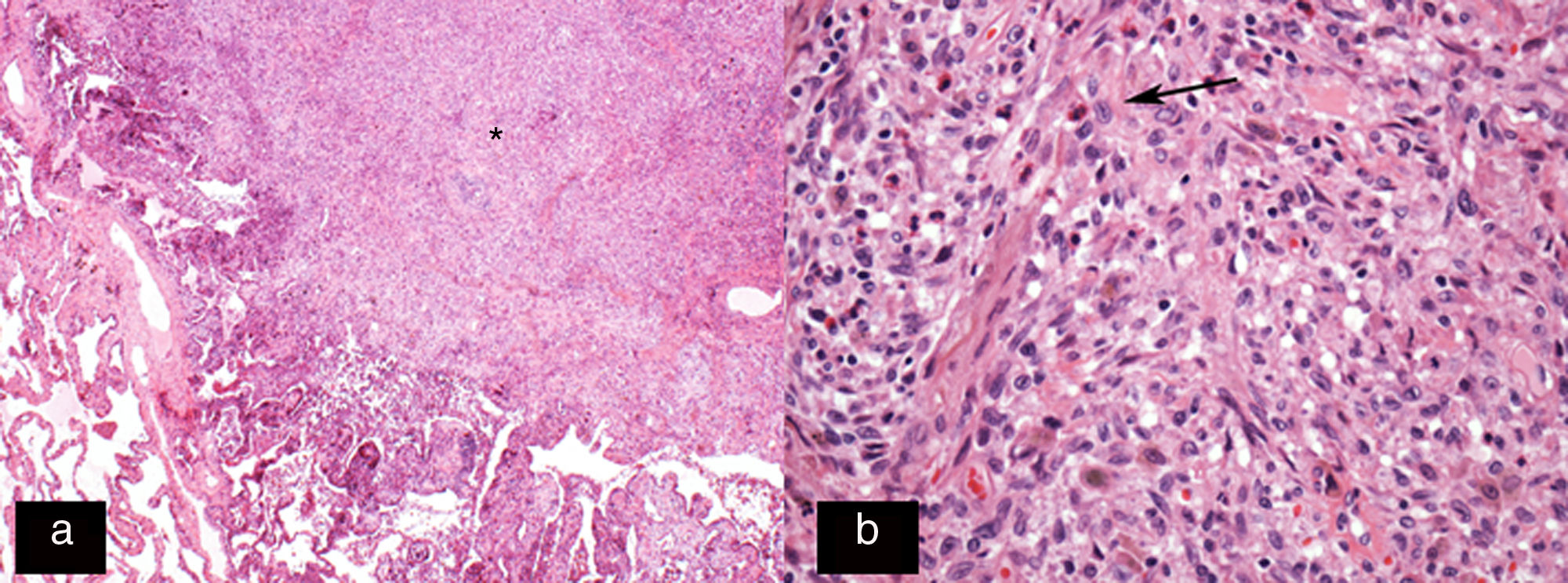
. (a) Star-shaped cellular nodule (asterisk). (b) Histological image at higher magnification (H–E 200×) showing the lesion to be predominantly composed of Langerhans cells (arrow) in combination with eosinophils and other inflammatory cells.")
Histology image stained with haematoxylin–eosin (H–E 20×). (a) Star-shaped cellular nodule (asterisk). (b) Histological image at higher magnification (H–E 200×) showing the lesion to be predominantly composed of Langerhans cells (arrow) in combination with eosinophils and other inflammatory cells.
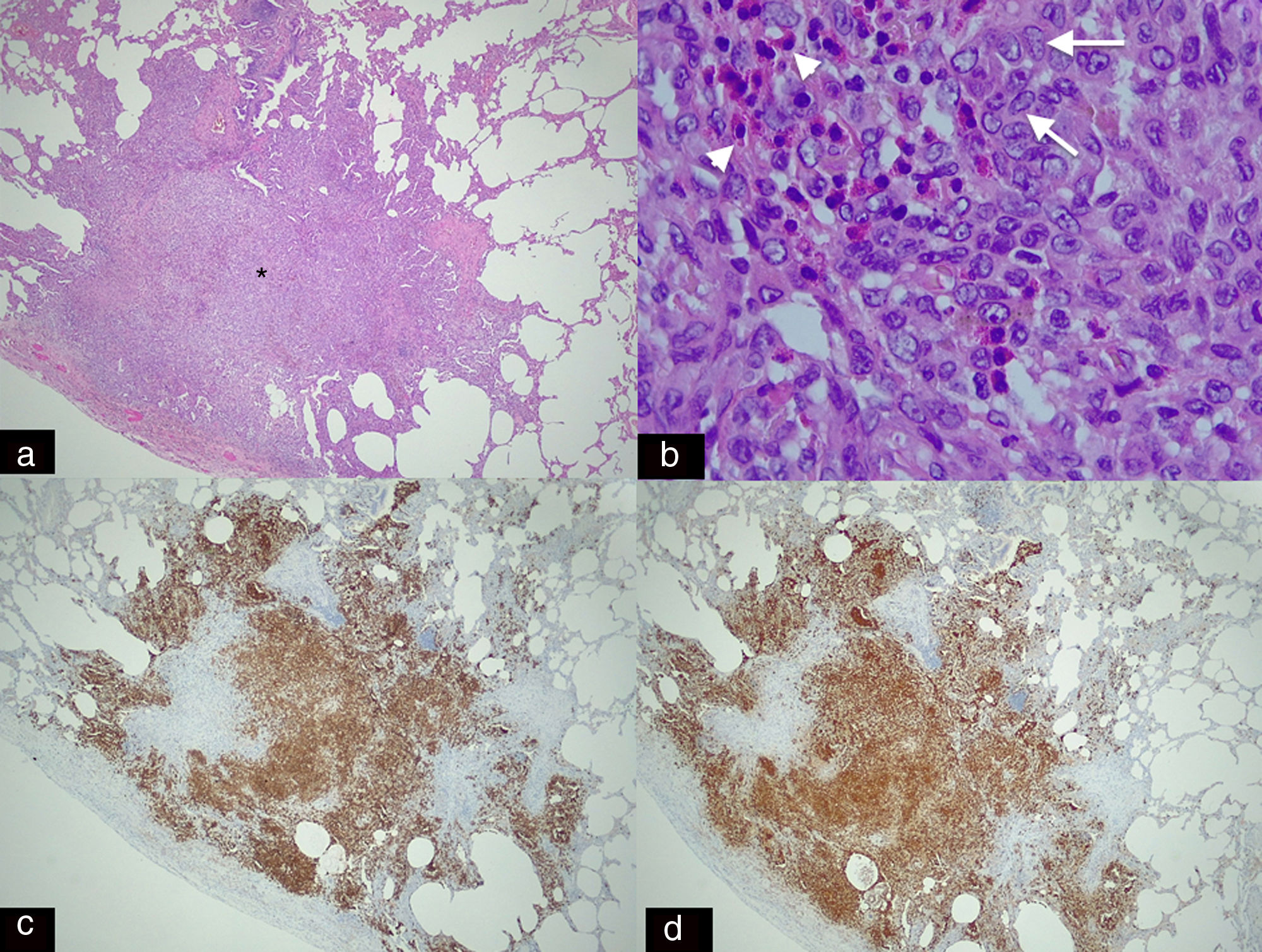
. (a) Spiculated nodule (asterisk) with irregular borders adjacent to a bronchus. This dense cellular aggregate is composed mainly of Langerhans cells and some eosinophils. (b) Higher magnification histological image (H–E 20×) showing infiltrates of Langerhans cells (arrows), characterised by their large size, oval shape, kidney-shaped nucleus and granular inclusion bodies (Birbeck granules) which are accompanied by eosinophilic infiltration (arrowheads). (c) Immunohistochemical staining for CD-1a (2.5×) and S-100 (2.5×). (d) Image showing extensive and intense cytoplasmic positivity in the Langerhans cells which confirms the diagnosis of Langerhans cell histiocytosis (same patient as in Fig. 5).")
Panoramic histological image stained with haematoxylin–eosin (H–E 2.5×). (a) Spiculated nodule (asterisk) with irregular borders adjacent to a bronchus. This dense cellular aggregate is composed mainly of Langerhans cells and some eosinophils. (b) Higher magnification histological image (H–E 20×) showing infiltrates of Langerhans cells (arrows), characterised by their large size, oval shape, kidney-shaped nucleus and granular inclusion bodies (Birbeck granules) which are accompanied by eosinophilic infiltration (arrowheads). (c) Immunohistochemical staining for CD-1a (2.5×) and S-100 (2.5×). (d) Image showing extensive and intense cytoplasmic positivity in the Langerhans cells which confirms the diagnosis of Langerhans cell histiocytosis (same patient as in Fig. 5).
Onset of LCH is usually when patients are in their 20s or 30s,12 with nonspecific symptoms such as dyspnoea, cough, fatigue and chest pain in the case of pneumothorax. However, up to a quarter of patients are asymptomatic.15 Recurrent pneumothorax is one of the most specific manifestations of this disease, although not the most common (10–25%).8,15
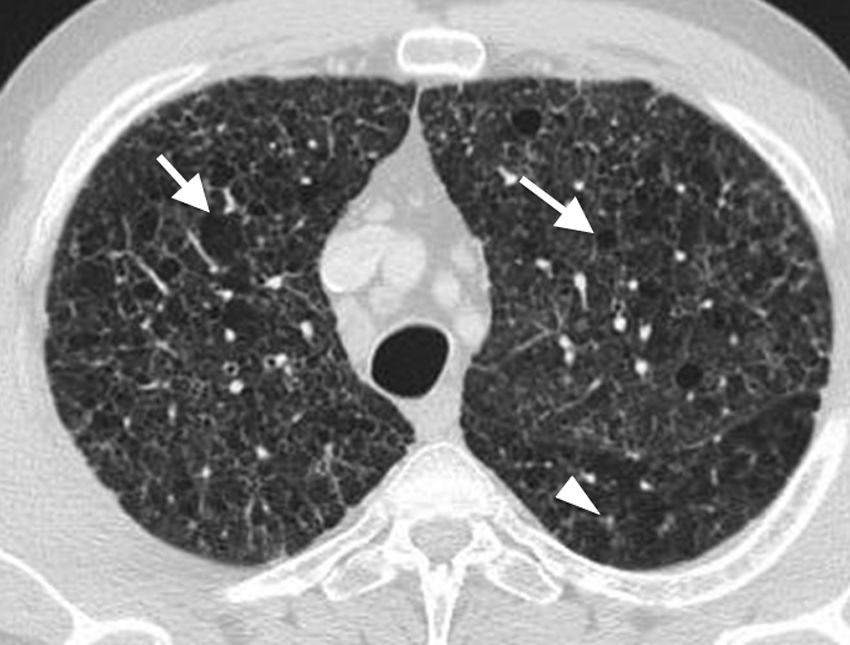
Imaging findingsIn the earliest stages, the disease is characterised by the presence of bilateral centrilobular nodules, predominantly in the upper and middle lung fields (Figs. 3 and 4).12 These initially smooth centrilobular nodules gradually become more star-shaped. Inflammation destroys the walls of the bronchiole, leaving the small airway dilated and providing the typical central radiolucency.8 The progressive dilation of the bronchioles is responsible for the appearance of cavitated nodules, with a progressively thinner wall (Figs. 4–6), irregularly shaped aerial cysts (Figs. 3 and 4) and, finally, confluence of these cysts.12 The costophrenic angles, as well as the anterior areas of the lung, are typically spared (Fig. 3).8
 showing confluent cysts of variable shape (arrows) and centrilobular nodules (arrowhead). These findings are typical of Langerhans cell histiocytosis.")
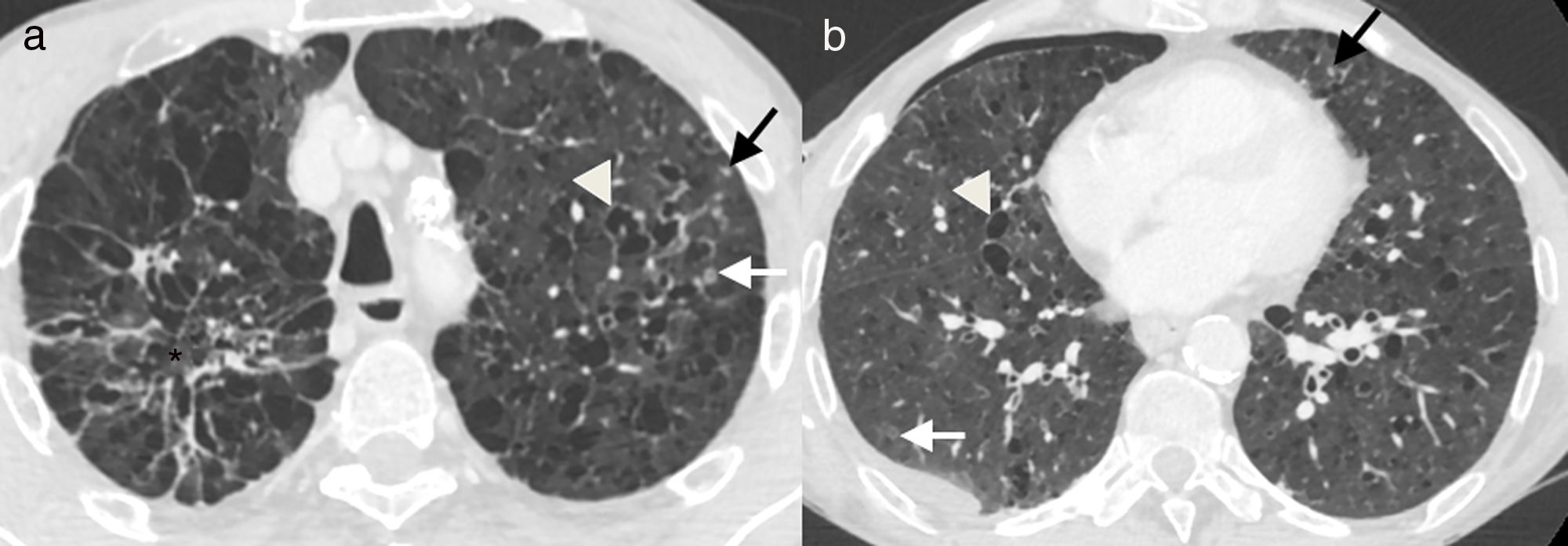
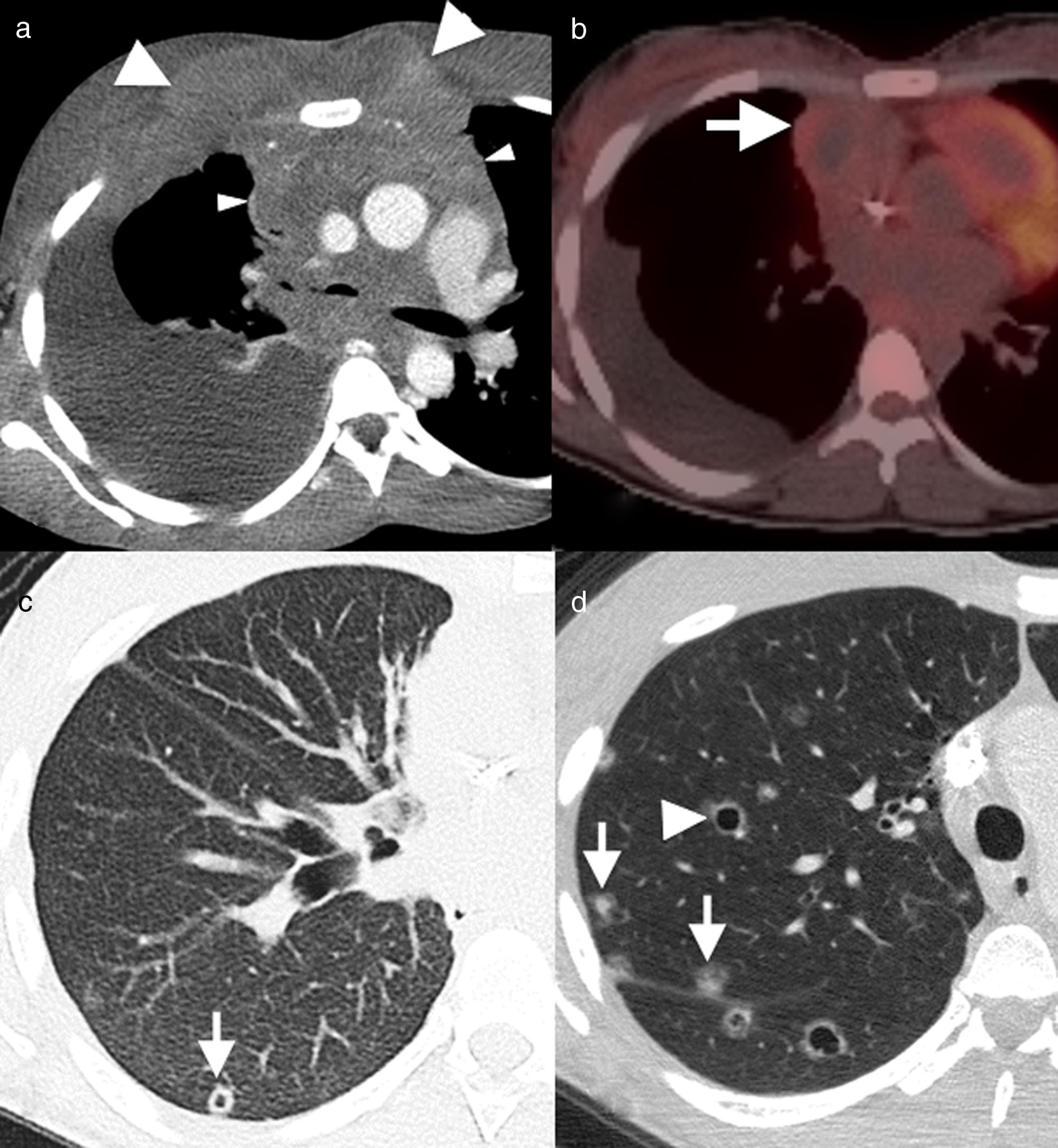
 Axial images of chest computed tomography. Small solid nodules (black arrows), some of them cavitated (white arrows), and multiple cysts of varying shape and size (arrowheads), associated with emphysema and areas of fibrosis (asterisk). There is a small right pneumothorax (b), which explains the clinical symptoms.")
55-Year-old male active smoker with acute chest pain. (a and b) Axial images of chest computed tomography. Small solid nodules (black arrows), some of them cavitated (white arrows), and multiple cysts of varying shape and size (arrowheads), associated with emphysema and areas of fibrosis (asterisk). There is a small right pneumothorax (b), which explains the clinical symptoms.
 Solid mass in the anterior mediastinum (asterisk), with loss of fat planes separating the vascular structures (arrows), suggesting vascular invasion. The patient had disease remission after chemotherapy, as seen in the axial image of positron emission tomography–computed tomography (PET–CT) (b). The mass has decreased in size and shows partial calcification, with no residual metabolic activity (arrow). Axial image in lung window (c) showing normal parenchyma. Follow-up CT, 12 months after the end of chemotherapy (d) showing multiple solid lung nodules (arrowheads) and cavitated (“Cheerio sign”) (arrows). The histological diagnosis confirmed histiocytic aggregates and Langerhans cells (Fig. 2) in this patient with Langerhans cell histiocytosis.")
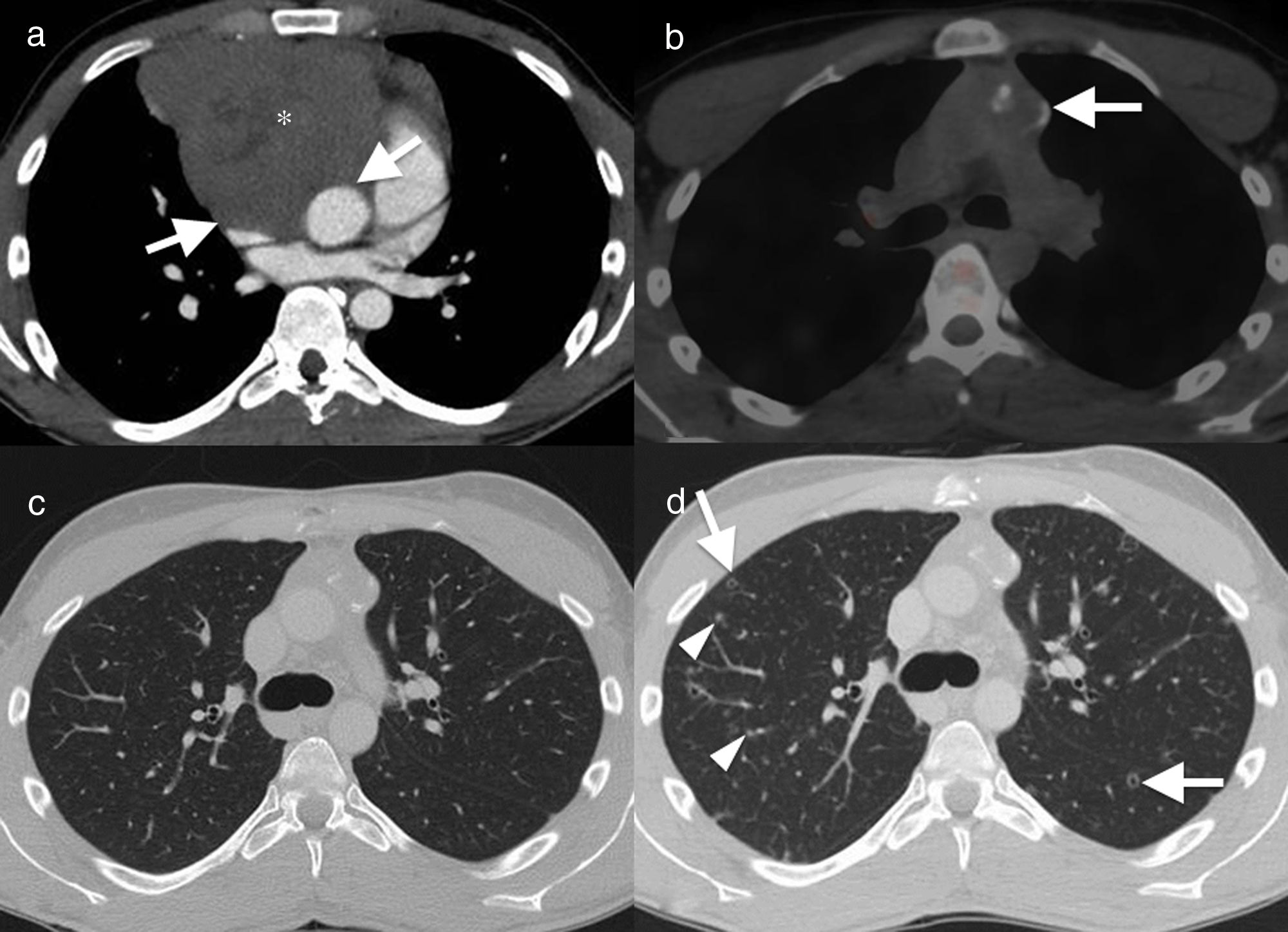
22-Year-old male non-smoker with a history of seminoma with extensive mediastinal involvement. Axial image of computed tomography with contrast. (a) Solid mass in the anterior mediastinum (asterisk), with loss of fat planes separating the vascular structures (arrows), suggesting vascular invasion. The patient had disease remission after chemotherapy, as seen in the axial image of positron emission tomography–computed tomography (PET–CT) (b). The mass has decreased in size and shows partial calcification, with no residual metabolic activity (arrow). Axial image in lung window (c) showing normal parenchyma. Follow-up CT, 12 months after the end of chemotherapy (d) showing multiple solid lung nodules (arrowheads) and cavitated (“Cheerio sign”) (arrows). The histological diagnosis confirmed histiocytic aggregates and Langerhans cells (Fig. 2) in this patient with Langerhans cell histiocytosis.

29-Year-old male non-smoker with Hodgkin's lymphoma. Axial image of chest computed tomography (CT), with contrast, in the initial diagnostic study (a) showing a solid mass (arrowheads) with involvement of multiple mediastinal compartments and infiltration in the anterior chest wall (arrows). (b) Axial image of positron emission tomography–CT, 12 months after the start of treatment, showing remission of the disease with the presence of a residual mass (arrow) with no abnormal metabolic activity. The infiltrative component in the anterior chest wall has been resolved. The CT image with lung window (c) shows one of the multiple cavitated lung nodules (arrow) present in the lung parenchyma. The possibility of opportunistic infection and lung metastases were considered in the light of this finding. In axial images of chest CT 2 months later (d), new multiple solid (arrows) and cavitated (arrowhead) lung nodules were detected bilaterally. The histology result ruled out metastatic disease and confirmed pulmonary Langerhans cell histiocytosis.
The findings described in the different stages very often overlap in the same patient. The term “Cheerio sign” has been used to describe the appearance of these lesions in their nodule stage with central radiolucencies, due to their similarity to ring-shaped cereals, and the main differential diagnosis is adenocarcinoma metastasis (Figs. 5 and 6d).16
Unlike in other interstitial diseases, lung volume remains normal or may be increased. Fibrosis can sometimes develop adjacent to the cysts and coexistence with emphysema is common.16
Pulmonary hypertension, also common in these patients, is associated with a worse prognosis. Computed tomography (CT) may show enlargement of the pulmonary arteries and, occasionally, ground-glass centrilobular nodules. As previously mentioned, some patients may present with pneumothorax (Fig. 4).8,9
The diagnosis of LCH is especially difficult in patients with a previous history of cancer, as, mainly in its nodular form, the radiological findings can be confused with metastatic disease or opportunistic infection. Other diagnostic considerations which have to be taken into account in any patient in its nodular form are miliary tuberculosis, sarcoidosis and silicosis.10
The differential diagnosis of cystic lung disease includes lymphangioleiomyomatosis, emphysema, interstitial lymphoid pneumonia, enlarged air spaces with fibrosis of the wall that may develop in the context of interstitial fibrosis related to smoking and bronchiectasis.10,12
It is important to remember that, although rare, LCH has to be included within the differential diagnosis of a diffuse nodular lung disease in the context of Hodgkin's lymphoma,1,8 particularly if it is associated with cysts and predominant involvement of the upper lobes and when there are no other signs of disease progression (Figs. 5 and 6).
TreatmentThe course of the disease is variable and unpredictable. In half of the patients, stopping smoking is sufficient for the disease to stabilise. In a quarter of the patients, the disease remits spontaneously, regardless of smoking cessation and, in the remaining patients, the disease progresses despite cutting down on smoking.8 In these cases where the disease progresses, additional measures are necessary, such as corticosteroid therapy or chemotherapy.10,17 Early intervention improves the prognosis, although in patients with advanced LCH, lung transplantation is considered as a therapeutic option.12
Rosai–Dorfman diseaseRDD, also known as sinus histiocytosis with massive lymphadenopathy, is a rare histiocytosis involving a benign proliferation of macrophages, in which LC are not involved.
Although RDD most typically involves the lymph nodes, there is sometimes lung involvement.
AetiopathogenesisThe aetiology is unknown but it is thought to be multifactorial. The herpes simplex virus and also macrophage colony-stimulating factor may have an influence on the aetiology of RDD, playing a role in the development and progression of the process. A relationship has been described with human immunodeficiency virus, amyloidosis, lymphoma and other lymphoproliferative processes,18 some autoantibodies, glomerulonephritis, Wiskott–Aldrich syndrome and polycythaemia vera.8
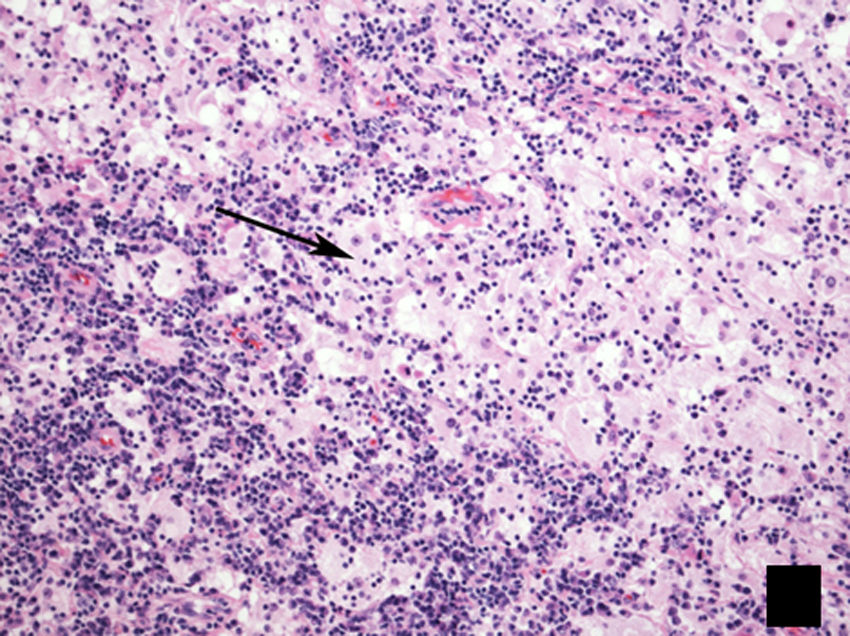
HistologyHistopathology study reveals an infiltrate of histiocytes, characterised by their pale eosinophilic cytoplasm, on a background of scattered lymphocytes and plasma cells. It is common to find emperipolesis (Fig. 7), which consists of phagocytosis of lymphocytes by histiocytes.19 Immunohistochemistry reveals the presence of S100-positive, CD68-positive, factor XIIIa-negative and CD1-negative cells.8
, showing the typical lesion present in Rosai–Dorfman disease, with a biphasic appearance composed predominantly of lymphocytes (dark areas) and histiocytes (light areas), showing emperipolesis (arrow).")
Histological image of lymph node biopsy, stained with haematoxylin–eosin (H–E 100×), showing the typical lesion present in Rosai–Dorfman disease, with a biphasic appearance composed predominantly of lymphocytes (dark areas) and histiocytes (light areas), showing emperipolesis (arrow).
RDD typically affects children and adolescents,8,10,18–20 and has a slight preference for males10 and individuals of African descent.10,18 The condition generally presents with painless, bilateral cervical lymphadenopathy (83%), pyrexia and weight loss.20 The form of the disease is extranodal in 20–43% of patients19: cutaneous (11.5%); nasal cavity (11.3%); upper aerodigestive tract (11.3%); orbital (8.5%); central nervous system (4.9%); renal (2.3%); hepatic and pancreatic. Chest involvement (2–3%) includes lymphadenopathy and lesion of the airway; the latter results in dyspnoea and spirometry alterations. Involvement of the lungs and pleura is rare.21
Imaging findingsIt is important to remember that chest involvement in RDD usually consists of mediastinal lymphadenopathy (Fig. 8). Involvement of the trachea and bronchi is usually in the form of single or multiple nodular masses of homogeneous density and with involvement of the adjacent fat, all of which lead to different degrees of airway obstruction.20
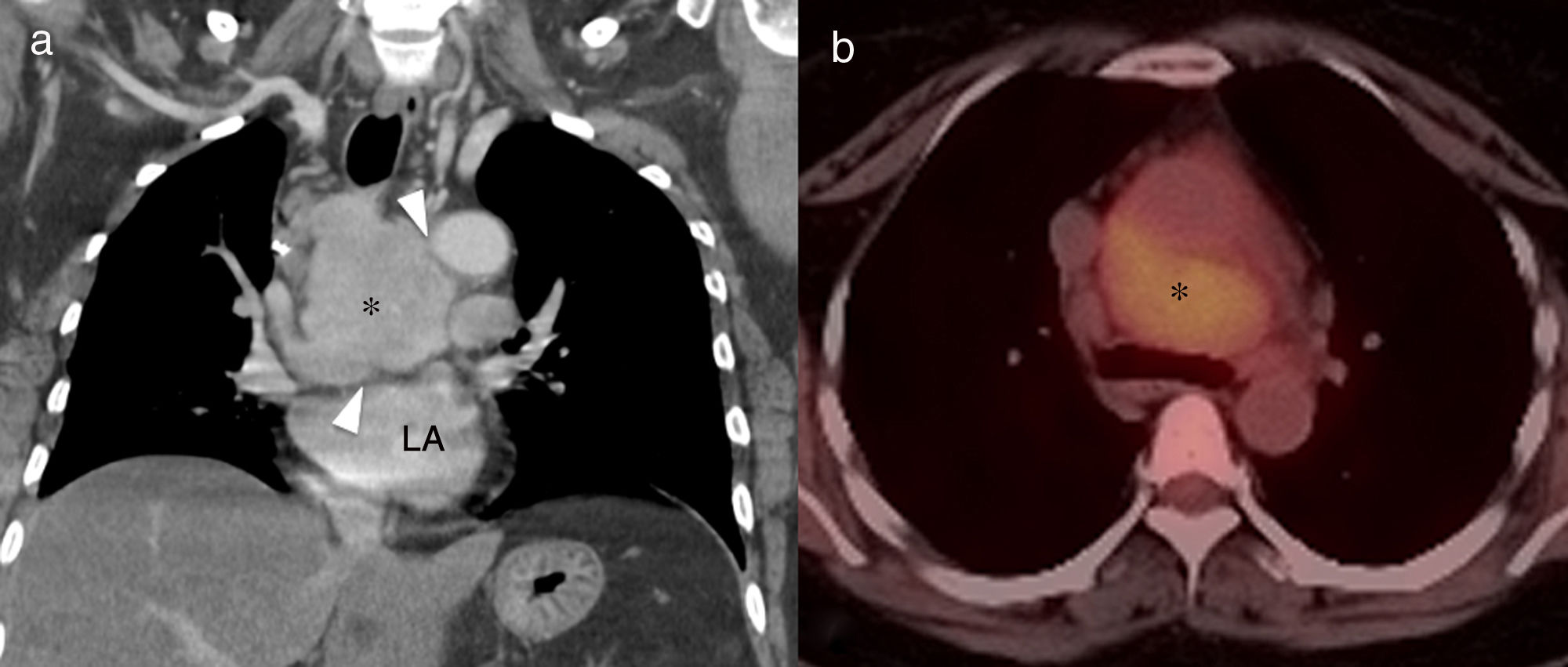
 reconstruction (a) showing a solid mediastinal mass with homogeneous enhancement (asterisk). The lesion is not affecting the fat planes separating the adjacent organs (arrowheads), and is metabolically active in positron emission tomography–CT (b). LA: left atrium.")
46-Year-old asymptomatic female with Rosai–Dorfman disease. Coronal computed tomography (CT) reconstruction (a) showing a solid mediastinal mass with homogeneous enhancement (asterisk). The lesion is not affecting the fat planes separating the adjacent organs (arrowheads), and is metabolically active in positron emission tomography–CT (b). LA: left atrium.
Lung involvement is characterised by poorly defined lung nodules or masses and perilymphatic interstitial infiltration, with no clear predominance of basal or apical fields. These lesions are generally associated with irregularity of the adjacent bronchi as a result of compression.18 Pleural involvement is also possible.8,18,19 The lesions are usually metabolically active in positron emission tomography (PET) (Fig. 8).18
When the disease manifests with mediastinal lymphadenopathy, the differential diagnosis includes mycobacterial infections, sarcoidosis, lymphoma, metastasis, immune reconstitution syndrome and fungal infections.
TreatmentThe majority of patients remain asymptomatic, with stable radiological findings, or spontaneous regression even occurs without the need for treatment. If patients have persistent symptoms, the response to corticosteroids is good. Chemotherapy has not been found useful.18
Erdheim–Chester diseaseECD is a rare histiocytosis, derived from dendritic cells other than LC. It is a systemic disease with heterogeneous manifestations and only a few hundred cases described in the literature.
AetiopathogenesisThe origin of ECD is unknown. In more than 50% of cases it is associated with BRAF V600E mutations in multipotent myelomonocytic precursor cells or tissue histiocytes.22
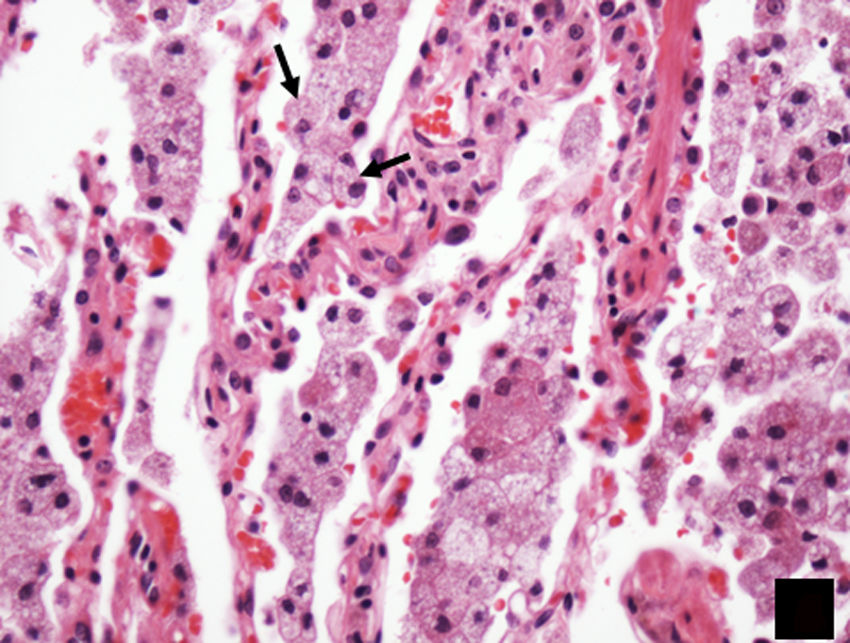
HistologyIt is characterised by perilymphatic xanthogranulomatous infiltrates, with foamy histiocytes surrounded by fibrosis.8 Immunocytochemistry is positive for CD68 and negative for CD1a. In contrast to LCH, the histiocytes have pale cytoplasm and do not have cytoplasmic eosinophilia or Birbeck granules (Fig. 9).
Signs and symptoms and typical of lung involvement due to Erdheim–Chester disease, showing alveolar spaces filled with foamy histiocytes (arrows).")
Onset of ECD is usually in middle age, and begins with bone pain secondary to metaphyseal and diaphyseal involvement in the long bones (50%).23 Extra-skeletal involvement may be cardiac, aortic, pulmonary, renal, CNS, orbital (manifesting exophthalmos) and cutaneous (xanthelasmas).24 Patients with lung (20–55%) and pleural involvement develop cough and progressive dyspnoea.8 Cardiac involvement can lead to conduction abnormalities or myocardial infarction if the coronary arteries become affected.23
The course of the disease essentially depends on its spread and distribution. Cardiac, pulmonary and neurological involvement are signs of poor prognosis.23 Cardiovascular manifestations are the main cause of death.23
Imaging findingsLung involvement is usually characterised by perilymphatic predominance, and thickening of the peribronchovascular interlobular septal interstitium (Fig. 10) and fissures are often found on CT.23 Ground-glass opacities23 and centrilobular nodules8,23 may also be seen. The presence of isolated cysts in some patients suggests a possible relationship with LCH; in fact, cases have been described in which both disorders coexist in the same patient (Fig. 11).24,25
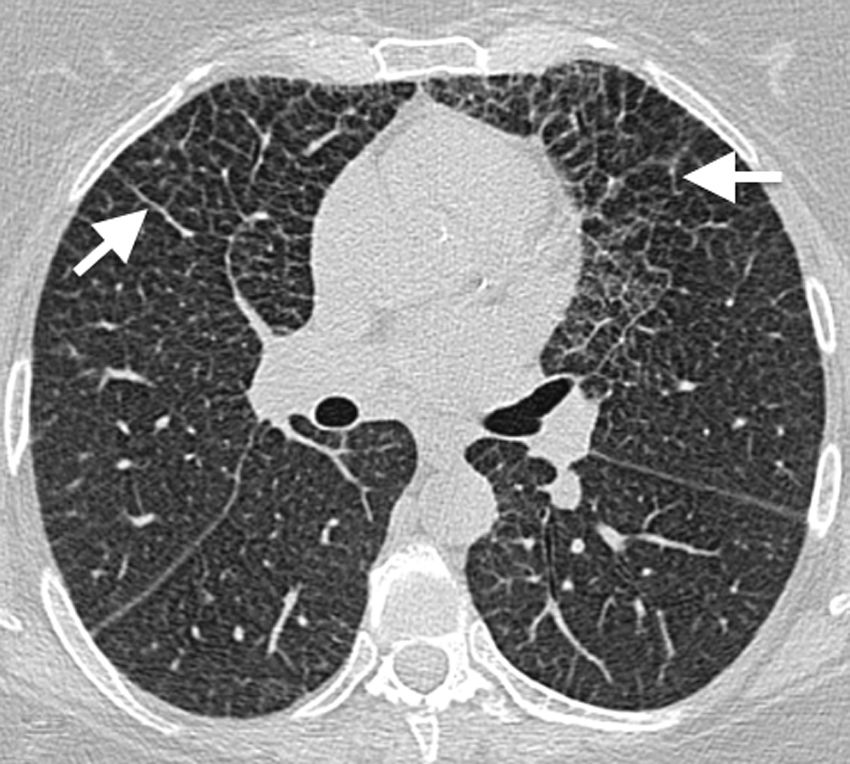
 image of the lung showing extensive cross-linking predominantly in anterior fields, with thickening of interlobular septa and intralobular interstitium (arrows). This is a classic manifestation of lung involvement in Erdheim–Chester disease.")
50-Year-old asymptomatic female with history of bilateral adrenal masses and splenomegaly. High resolution axial computed tomography (CT) image of the lung showing extensive cross-linking predominantly in anterior fields, with thickening of interlobular septa and intralobular interstitium (arrows). This is a classic manifestation of lung involvement in Erdheim–Chester disease.
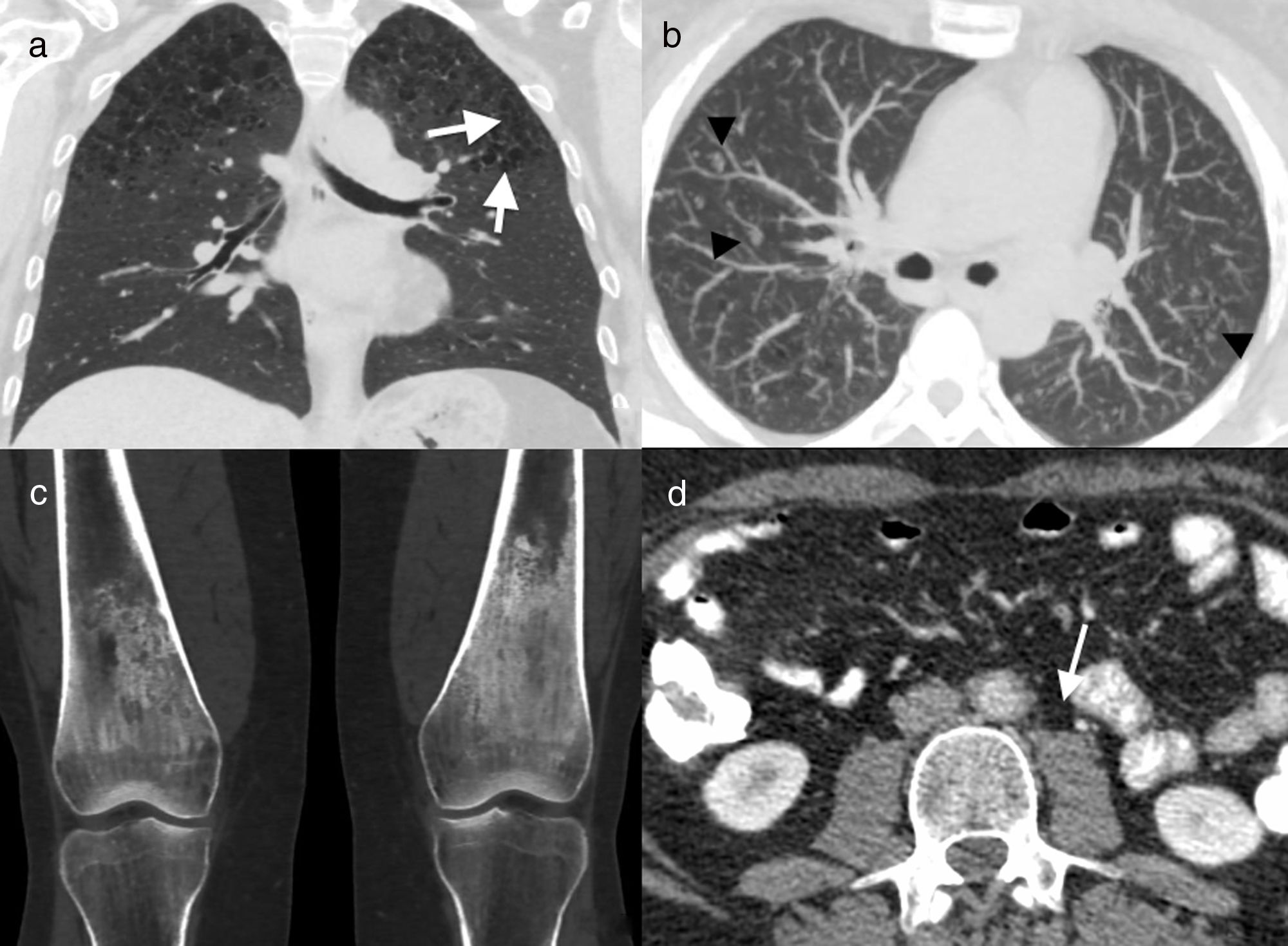
 coronal reconstruction (a) and axial image with maximum intensity projection (MIP) (b). Cysts (arrows) and multiple centrilobular nodules (arrowheads) in both upper lobes, together with centriacinar emphysema. Coronal CT reconstruction of knees (c) showing symmetric bilateral sclerosis in the diaphysis and metaphysis of both femurs. Axial image of abdominal CT (d) in which concentric thickening of the wall of the abdominal aorta can be seen (arrow). Biopsy of a mandibular lesion (not illustrated) confirmed the diagnosis of Erdheim–Chester disease.")
41-Year-old male former smoker with Erdheim–Chester disease. Computed tomography (CT) coronal reconstruction (a) and axial image with maximum intensity projection (MIP) (b). Cysts (arrows) and multiple centrilobular nodules (arrowheads) in both upper lobes, together with centriacinar emphysema. Coronal CT reconstruction of knees (c) showing symmetric bilateral sclerosis in the diaphysis and metaphysis of both femurs. Axial image of abdominal CT (d) in which concentric thickening of the wall of the abdominal aorta can be seen (arrow). Biopsy of a mandibular lesion (not illustrated) confirmed the diagnosis of Erdheim–Chester disease.
Patients may develop pleural thickening and pleural effusion. Pleural thickening is usually located in the right paravertebral basal area and it can at times continue with infiltration of the retrocrural space, creating a pseudotumour-like mass with soft tissue density.24
Cardiovascular involvement is common and usually affects the descending and abdominal aorta, with periadventitial infiltration of soft tissue density (“coated aorta” appearance) which can extend to the supra-aortic trunks or intercostal or coronary arteries, simulating vasculitis.11,24 Takayasu's disease and retroperitoneal fibrosis are the main differential diagnoses.
Infiltration of the wall of the right atrium with a pseudotumour appearance may also be seen.24 When the mediastinal infiltration is extensive and compresses the vascular structures, the differential diagnosis includes: chronic granulomatous disease, lymphoma, fibrosing mediastinitis and amyloidosis.
It is important to remember that, due to the non-specific nature of the pulmonary involvement in ECD, extra-thoracic manifestations are usually key in the diagnosis. The presence of sclerotic lesions in the metaphysis and diaphysis of long bones (Fig. 12) and the typical renal sign “hairy kidney” (infiltration of contrast-enhanced perirenal fat)22 (Fig. 12) support the diagnosis.
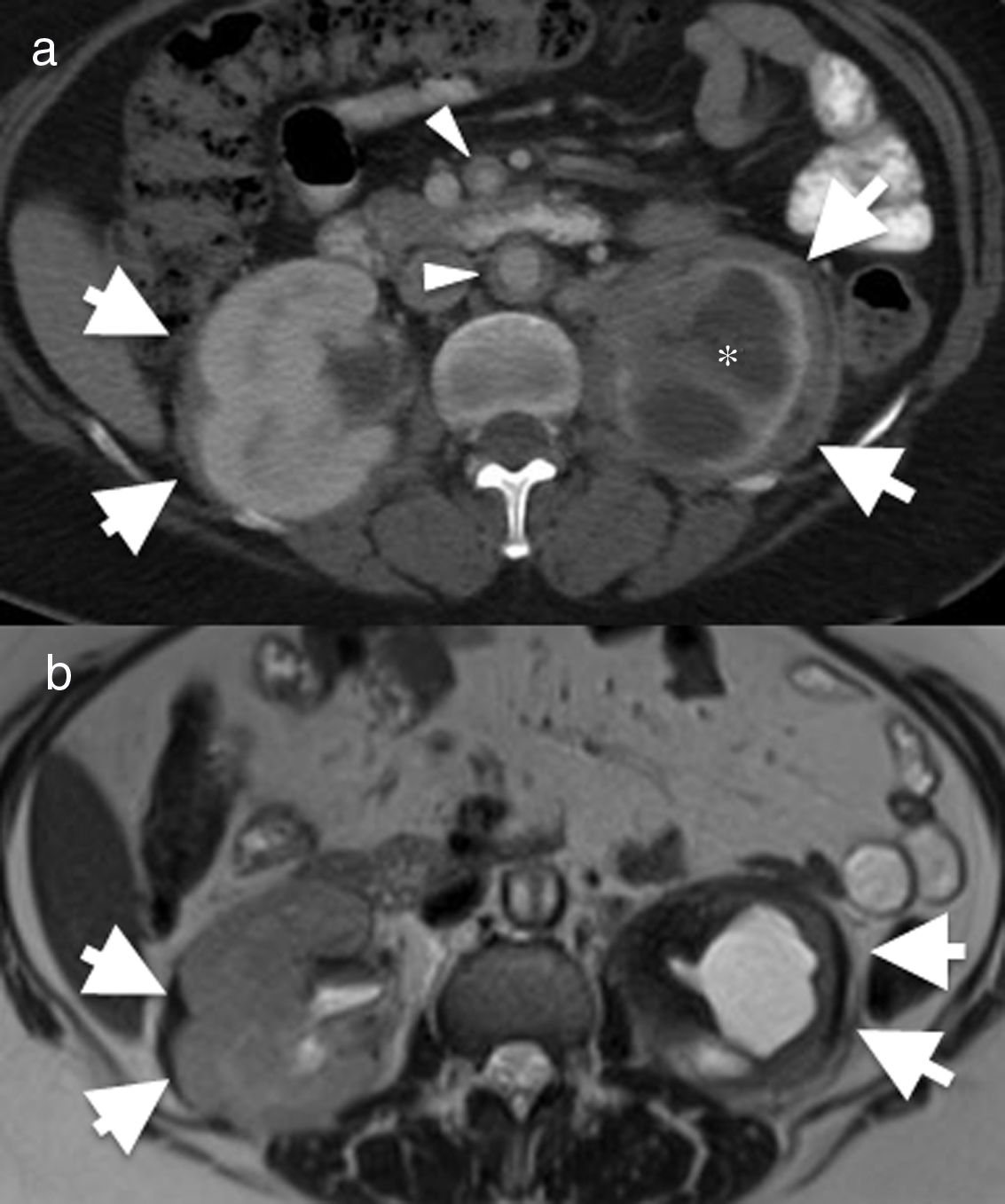
 image of the abdomen (a) showing bilateral perirenal infiltration by soft tissue (arrows). There is significant atrophy of the left kidney and severe hydronephrosis (asterisk). There is also mural thickening in the abdominal aorta and superior mesenteric artery (arrowheads). (b) Axial magnetic resonance imaging (MRI) T2-weighted spin echo showing the low signal intensity of the perirenal soft tissue (arrows) and of the periaortic infiltrate (arrowhead). The differential diagnosis with this finding includes Erdheim–Chester disease, retroperitoneal fibrosis, sarcoidosis and amyloidosis. The biopsy result confirmed Erdheim–Chester disease. This patient had no disease-related lung involvement.")
55-Year-old female with Erdheim–Chester disease. She initially presented with lower left quadrant abdominal pain and nausea. Axial computed tomography (CT) image of the abdomen (a) showing bilateral perirenal infiltration by soft tissue (arrows). There is significant atrophy of the left kidney and severe hydronephrosis (asterisk). There is also mural thickening in the abdominal aorta and superior mesenteric artery (arrowheads). (b) Axial magnetic resonance imaging (MRI) T2-weighted spin echo showing the low signal intensity of the perirenal soft tissue (arrows) and of the periaortic infiltrate (arrowhead). The differential diagnosis with this finding includes Erdheim–Chester disease, retroperitoneal fibrosis, sarcoidosis and amyloidosis. The biopsy result confirmed Erdheim–Chester disease. This patient had no disease-related lung involvement.
The recent discovery of activating mutations in the BRAF gene has led to the development of new therapies focused on inactivation of the gene. Such therapies are currently reserved for cases resistant to treatment, when first-line therapies (corticosteroids, immunosuppressants, chemotherapeutic agents) are not effective, alone or in combination with the first-line treatment. The new therapies include: vemurafenib (a BRAF inhibitor); infliximab (an anti-TNF antibody); and anakinra (an interleukin 1 receptor antagonist). Even so, five-year survival is no more than 70%.22
ConclusionLCH, RDD and ECD are a set of disorders with idiopathic causes, in which the proliferation and infiltration of histiocytes in the histological samples is the diagnostic pathology finding. When these disorders affect the lung, with the exception of LCH, the findings become fairly non-specific and, occasionally, the extrapulmonary manifestations provide the diagnostic key. Although very rare, LCH should be considered in non-smoking patients on treatment with chemotherapy and radiotherapy with new onset of cavitated nodules, and included in the differential diagnosis along with metastatic disease and opportunistic infection.
Authorship- 1.
Responsible for the integrity of the study: CTG, JB, EC, EBS, NM and LF.
- 2.
Study conception: CTG, JB and LF.
- 3.
Study design: CTG, JB, EC, EBS, NM and LF.
- 4.
Data acquisition: CTG, JB, EC, EBS, NM and LF.
- 5.
Analysis and interpretation of the data: N/A.
- 6.
Statistical processing: N/A.
- 7.
Literature search: CTG, JB and LF.
- 8.
Drafting of the paper: CTG, JB and LF.
- 9.
Critical review of the manuscript with relevant intellectual contributions: CTG, JB, EC, EBS, NM and LF.
- 10.
Approval of the final version: CTG, JB, EC, EBS, NM and LF.
The authors declare that they have no conflicts of interest.
Please cite this article as: Trejo Gallego C, Bueno J, Cruces E, Stelow EB, Mancheño N, Flors L. Histiocitosis pulmonar: más allá de la histiocitosis de células de Langerhans relacionada con el tabaco. Radiología. 2019;61:215–224.

