












Supplement “Pulmonary Interstitial Pathology”
More infoExposure to smoke is associated with the development of diseases of the airways and lung parenchyma. Apart from chronic obstructive pulmonary disease (COPD), in some individuals, tobacco smoke can also trigger mechanisms of interstitial damage that result in various pathological changes and pulmonary fibrosis. A causal relation has been established between tobacco smoke and a group of entities that includes respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), desquamative interstitial pneumonia (DIP), Langerhans cell histiocytosis (LCH), and acute eosinophilic pneumonia (AEP). Smoking is considered a risk factor for idiopathic pulmonary fibrosis (IPF); however, the role and impact of smoking in the development of this differentiated clinical entity, which has also been called combined pulmonary fibrosis and emphysema (CPFE) as well as nonspecific interstitial pneumonia (NIP), remains to be determined. The definition of smoking-related interstitial fibrosis (SRIF) is relatively recent, with differentiated histological characteristics. The likely interconnection between the mechanisms involved in inflammation and pulmonary fibrosis in all these processes often results in an overlapping of clinical, radiological, and histological features in the same patient that can sometimes lead to radiological patterns of interstitial lung disease that are impossible to classify. For this reason, a combined approach to diagnosis is recommendable. This combined approach should be based on the joint interpretation of the histological and radiological findings while taking the clinical context into consideration. This paper aims to describe the high-resolution computed tomography (HRCT) findings in this group of disease entities in correlation with the clinical manifestations and histological changes underlying the radiological pattern.
La exposición al tabaco guarda una relación reconocida con el desarrollo de enfermedades de la vía aérea y el parénquima pulmonar. Aparte de la enfermedad pulmonar obstructiva crónica, en algunos individuos el humo del tabaco puede desencadenar mecanismos de daño intersticial que resultan en variadas alteraciones patológicas y fibrosis pulmonar. Se ha reconocido un grupo de entidades con una relación causal establecida con el tabaco, que incluye la bronquiolitis respiratoria con enfermedad pulmonar intersticial, la neumonía intersticial descamativa, la histiocitosis de células de Langerhans y la neumonía eosinófila aguda. Aunque el tabaco se considera factor de riesgo, aún es ambiguo su papel e impacto en el desarrollo de la fibrosis pulmonar idiopática, la entidad clínica diferenciada que se ha denominado combinación fibrosis pulmonar y enfisema, y la neumonía intersticial no específica. La definición de la fibrosis intersticial asociada al tabaco es relativamente reciente, con características histológicas diferenciadas. La interconexión probable entre los mecanismos que determinan la inflamación y la fibrosis pulmonar en todos estos procesos referidos se traduce con frecuencia en superposición de rasgos histológicos, clínicos y radiológicos en el mismo paciente, que en algunos casos puede determinar patrones radiológicos de neumopatía intersticial no clasificables. Por ello es recomendable un abordaje combinado para el diagnóstico, que debe estar basado en la interpretación conjunta de las características histológicas y hallazgos radiológicos, en el contexto clínico apropiado. Nuestro objetivo se centra en la descripción de los hallazgos radiológicos en la tomografía computarizada de alta resolución, en correlación con las manifestaciones clínicas y las alteraciones histológicas subyacentes al patrón radiológico.
The relationship between smoking and a broad group of interstitial lung diseases is increasingly recognised, with a significant tendency towards overlapping and co-existing interstitial lung injury and emphysema patterns.
HRCT plays a prominent role in diagnosis and many cases, averts the need for lung biopsy. However, radiological patterns reflect this overlapping of histological patterns, which is why they are often mixed and non-specific and often difficult to classify.
SRIF (smoking-related interstitial fibrosis) is a defined histological picture, different from other ILDs, with little or no clinical relevance. However, it is widespread in lung biopsies in smokers.
CPFE (combined pulmonary fibrosis and emphysema) is increasingly recognised as a highly symptomatic clinical syndrome associated with a poor prognosis in heavy smokers.
The need for a multidisciplinary clinical, histological and radiological combined approach is justified, with the aim of recognising those patterns that may have greater clinical relevance, including those associated with fibrosis with the likelihood of progression, as well as determining the need for biopsy and its most suitable location.
IntroductionThe potential causal relationship between tobacco exposure and lung cancer, chronic bronchial disease, and emphysema is well known, as is its role as a risk factor for cardiovascular disease and stroke. The relationship between smoking and diffuse interstitial lung disease (ILD) is less well understood but is also widely accepted. With the expansion of programmes for the early detection of lung cancer, the detection of subclinical or incidental interstitial changes has increased significantly.1,2 In a large-scale prospective cohort study, it has been demonstrated that 8% of smokers have interstitial lung abnormalities on high-resolution computed tomography (HRCT). These abnormalities are associated with an increased relative risk of all-cause mortality 3 and some progress to pulmonary fibrosis.4
Smoking-related ILD comprises a heterogeneous set of disorders, the histological manifestations of which range from reversible inflammatory changes to emphysema and fibrosis. These diseases can be classified into two groups. The first would include those whose causal relationship with smoking has traditionally been established: desquamative interstitial pneumonia (DIP), respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), Langerhans cell histiocytosis (LCH),5 and acute eosinophilic pneumonia (AEP), which has been associated with changes in smoking habits. The second group covers a series of entities in which smoking is implicated as a probable risk factor in conjunction with other endogenous and exogenous factors. This group includes idiopathic pulmonary fibrosis (IPF), the distinct clinical entity that has been called combined pulmonary fibrosis and emphysema (CPFE), and non-specific interstitial pneumonia (NSIP).1,6 Smoking-related interstitial fibrosis (SRIF) has been defined as a histological picture with characteristics that differentiate it from IPF and other interstitial diseases.7,8
Although most of these disorders have been considered individual entities, the possible interconnectedness of the mechanisms that cause inflammation and pulmonary fibrosis in smokers results in a frequent overlap of histological patterns5,9–11 and coexistence with emphysema, which is a much more common consequence of smoking. This translates into radiological patterns, which in some cases may be unclassifiable.12 However, despite the difficulties involved in diagnosing these diseases, not only from the histological and radiological point of view but also clinically, it is essential to define this diagnosis well, and especially to differentiate usual interstitial pneumonia (UIP) and other diffuse fibrosing lung diseases of entities that have a better prognosis, as is the case with SRIF.8,13 Hence the advisability to approach the diagnosis in a multidisciplinary way for a better understanding and differentiation of these disorders.
In this review, we will describe the clinical, histological, and radiological characteristics of interstitial diseases with an established or suspected causal relationship with smoking, paying particular attention to the translation of histopathological manifestations on HRCT and the challenges posed to the radiologist in the interpretation of these findings.
Pulmonary Langerhans cell histiocytosisPulmonary LCH (PLCH) is the pulmonary form of presentation of LCH. LCH is usually restricted to the lung, although it is estimated that in 5%–15% of patients, it affects multiple organs.14 Ninety percent of patients with PLCH are smokers or ex-smokers. It has no predilection regarding sex and occurs most frequently in a person's 20s or 30s. Between 10% and 25% of patients are asymptomatic and the rest present with non-specific symptoms, such as cough or exertional dyspnoea, although fever, weight loss or anorexia may also appear.1,11 In 15% of cases, the form of presentation is a spontaneous pneumothorax, which can be recurrent (Fig. 1d and f).1 In advanced cases, pulmonary hypertension (PHTN) is relatively common, and is more prominent than in other chronic lung diseases.15
 reveal a greater profusion of cavitated and non-cavitated pulmonary nodules and cysts (blue arrows). In those from 2011 (images d–f), cysts (blue arrows) are observed, some of very variegated morphology, which were complicated by a left pneumothorax (orange arrow). The cysts leave the lung bases, and the costophrenic angles preserved and are less numerous in the anterior areas.")
Patient diagnosed with PLCH, smoker of three packs/day. Axial HRCT slices and coronal reconstruction. Images from 2010 (a–c) reveal a greater profusion of cavitated and non-cavitated pulmonary nodules and cysts (blue arrows). In those from 2011 (images d–f), cysts (blue arrows) are observed, some of very variegated morphology, which were complicated by a left pneumothorax (orange arrow). The cysts leave the lung bases, and the costophrenic angles preserved and are less numerous in the anterior areas.
Its aetiopathogenesis is unknown. Langerhans cells (LCs) are a type of dendritic antigen-presenting cell found primarily in the skin and mucosa. They have a myeloid origin and in some conditions they can be recruited to the lung. Tobacco smoke appears to play an important role in this differentiation, although the pathophysiological mechanism by which it acts as a triggering factor is unknown. It is hypothesised that LCH may represent a myeloid neoplastic proliferation because the expansion of the LCs is clonal. This hypothesis is supported by the association described between LCH and some neoplasms, especially haematological ones,1,16 and the recent discovery of some associated oncogenic mutations. These findings raise the debate about the neoplastic versus inflammatory origin of the pathophysiological mechanisms triggered by smoking in the development of LCH.1
The histopathological picture is characterised by well-individualised bronchiolocentric interstitial stellate nodules, which in the initial stage are cellular, composed of LCs and a variable proportion of lymphocytes, fibroblasts, eosinophils, neutrophils, plasma cells and pigmented macrophages. LCs have a characteristic lobulated and cleft nucleus, in addition to CD1a, and CD207 (langerin), as immunohistochemical markers on the cell surface and positivity for the cytoplasmic protein S-100. These markers can contribute to the diagnosis using bronchoalveolar lavage (BAL), although it is a technique with low sensitivity.1,11 Over time, the cellularity of the initial nodules is centripetally replaced by fibrotic tissue, forming mixed nodules that, in the final stage, transform into bronchiolocentric stellate scars containing only fibrotic tissue, surrounded by enlarged and distorted air spaces11 (Fig. 2).
 with an infiltrate made up of anthracotic macrophages and scanty Langerhans cells, and traction emphysema in the periphery (H&E 20⋅ and 10⋅).")
Lung biopsy with findings of PLCH in the fibrotic stage. In a and b, at higher and lower magnification, respectively, a nodular-stellate fibrotic lesion (orange arrows) with an infiltrate made up of anthracotic macrophages and scanty Langerhans cells, and traction emphysema in the periphery (H&E 20⋅ and 10⋅).
The HRCT findings (Fig. 1a–f) also vary with the evolution of the disease, in parallel with the histological findings. In the initial phases, centrilobular nodules of one to 10mm appear, which cavitate as the disease progresses, finally becoming thin-walled cysts. In the intermediate phases, the characteristic pattern in HRCT is a combination of nodules and cysts with walls of intermediate thickness, generally of irregular morphology, which predominate in the upper and middle lung fields, with the costophrenic recesses and the anterior zones of the lung preserved11,14 In more advanced stages, the predominance of irregular thin-walled cysts that can measure up to 20mm in diameter is typical. Lung volumes are usually preserved or increased. Reticulation, emphysema, and areas of fibrocystic distortion may coexist.17 Ground-glass opacities may also appear, which correlate with the presence of RB and DIP histological changes, supporting the idea that PLCH, RB, and DIP comprise a spectrum of lung injury in smokers.5
The differential diagnosis must be established with lymphangioleiomyomatosis, in which the cysts have a more regular, rounded morphology and adopt a diffuse distribution without the lung bases being preserved; with centrilobular emphysema (CLE) in the most advanced stages of the disease; with lymphocytic interstitial pneumonia or Birt-Hogg-Dubé syndrome. In the more nodular or mixed forms, metastases, infections, tracheobronchial papillomatosis, or sarcoidosis could be considered.18,19
Transbronchial biopsy has a diagnostic yield of 50%. However, surgery usually provides a definitive diagnosis.1
PLCH remits spontaneously in 25% of patients, even if tobacco use persists. Quitting smoking stabilises the disease in 50% of cases, although in the remaining cases, it can progress despite this. Despite no specific pharmacological treatment, corticosteroids, immunosuppressants, and chemotherapy have been tried with mixed results. In the case of lung transplantation, PLCH can recur in up to 20% of cases.1
Respiratory bronchiolitis-associated interstitial lung diseaseRB is a very common histopathological finding in smokers, with most patients being asymptomatic. It is characterised by an accumulation of pigmented macrophages in the terminal bronchioles20 (Fig. 3).
 HRCT apical axial slice with faint centrilobular nodules (blue arrow). (b) Histological section with RB data: terminal bronchiole with intraluminal pigmented macrophages (orange arrow) (H&E 20⋅).")
RB-ILD in a smoker. A 59-year-old woman who smokes a pack a day, without respiratory symptoms, with symptoms of asthenia. (a) HRCT apical axial slice with faint centrilobular nodules (blue arrow). (b) Histological section with RB data: terminal bronchiole with intraluminal pigmented macrophages (orange arrow) (H&E 20⋅).
The differentiation between RB and RB-ILD is established according to the presence or absence of respiratory symptoms. Patients with RB-ILD have respiratory symptoms and are usually heavy smokers. The histological distinction between the two is controversial; according to some authors, BR-EPI tends to present greater alveolar fibrosis.21
HRCT findings include centrilobular nodules and irregular ground-glass opacities (Fig. 3a) and central thickening of the bronchial walls, predominantly in the upper lobes in some series. In addition to CLE, areas of air trapping and ground-glass opacities are more common in heavy smokers, potentially reversible signs if the patient quits smoking.22,23 This entity is diagnosed without the need for surgical biopsy in smokers with the characteristic findings described in HRCT and with the finding of pigmented macrophages and absence of lymphocytosis in the BAL since the presence of lymphocytosis in the BAL is a more characteristic finding of hypersensitivity pneumonitis (HP).11,24 HP also presents with centrilobular nodules on HRCT, so the radiological differential diagnosis must also be established with it.25 Some authors have argued a supposed protective effect of tobacco against HP.16
The most important recommendation in these patients is that they give up smoking. However, clinical and physiological improvement is slow and incomplete.26,27
Desquamative interstitial pneumoniaDIP was first described by Liebow et al.28 in 1965. The term DIP is still used, although it has been demonstrated that there is no "desquamation of epithelial cells", but an abnormal accumulation of pigmented macrophages in the alveolar spaces, so it is thought to be a misnomer.1
It is one of the rarest forms of idiopathic interstitial pneumonia and is also one of the smoking-related interstitial diseases.29 RB and DIP are thought to be part of the same disease spectrum. Although it is accepted that smoking is a risk factor for DIP, the relationship between the two is not as strong as it is in RB-ILD. DIP has occasionally been described in non-smokers in relation to environmental factors, exposure to dust or drugs.11,21,23
The disease presents insidiously between in a person's 30s to 40s, with symptoms similar to RB-ILD, although in general, with more severe functional limitation, dyspnoea, and dry cough. Acropachy occurs in almost half of patients.24,30
DIP has a worse prognosis than does RB-ILD. The most important recommendation is to give up smoking. However, according to some series, clinical and functional improvement is often late and incomplete and occurs in the long term in 28% and 10.5% of cases.27
The most striking histopathological finding of DIP is the deposition of pigmented macrophages in the alveolar spaces31 (Fig. 4d). The alveolar septa may be thickened by inflammation and even fibrosis, which, although mild, is generally more severe than in RB. The alveolar architecture is usually preserved, and honeycombing is scanty or absent. The histological characteristics are similar to those of RB-ILD, and the key to differentiating these two entities is the distribution and extension of the lesions, which would be bronchiolocentric in RB-ILD and diffuse in DIP.11 BAL in these patients contains a large number of pigmented macrophages.
 revealing diffuse ground-glass pulmonary opacities (blue arrows), more extensive in the middle and basal areas, in some locations presenting with some associated central bronchiectasis. Histological section revealing pigmented alveolar macrophages (d) (orange arrow) in areas of emphysema and minimal subpleural interstitial fibrosing component (H&E 10⋅).")
Male smoker with DIP. Axial HRCT slices (images a–c) revealing diffuse ground-glass pulmonary opacities (blue arrows), more extensive in the middle and basal areas, in some locations presenting with some associated central bronchiectasis. Histological section revealing pigmented alveolar macrophages (d) (orange arrow) in areas of emphysema and minimal subpleural interstitial fibrosing component (H&E 10⋅).
The main radiological findings on HRCT include diffuse irregular ground-glass opacities, which can be bilateral, with a basal and peripheral predominance (Fig. 4). In the evolution of the disease, the appearance of small cysts within the ground-glass areas has been described. However, the appearance of reticulation and honeycombing —which would reflect fibrosis— is unusual. Cysts have been associated with dilated bronchioles and alveolar ducts on HRCT.32,33 In follow-up of treated patients with HRCT, the ground-glass opacities should progressively decrease or disappear.33
The radiological differential diagnosis includes RB-ILD. A radiopathological relationship has been described between macrophage occupation of the respiratory bronchioles and centrilobular nodules on HRCT. In patients who also present with ground-glass opacities (Fig. 3a), we may think that there is an overlap with DIP, since the radiopathological relationship between the occupation of the alveoli and alveolar sacs by macrophages and the ground-glass opacities has also been described on HRCT.22,26 Other differential diagnoses include NSIP and atypical infections, such as Pneumocystis jirovecci pneumonia.11,24,25 The relationship between DIP and NSIP is unclear.11,29,33 The difficulties posed by the distinction between the two are discussed in the section corresponding to NSIP.
Acute eosinophilic pneumoniaAEP is a rare disease mainly affecting males from their teens to their 30s. At least two thirds of patients are smokers.34–38 Although the cause is, in many cases, unknown, a direct relationship between a change in smoking habits and the triggering of this disease has been reliably demonstrated regarding starting smoking, smoking more or re-starting smoking after quitting.39,40
AEP is a rapidly progressive acute disease with fever, dyspnoea, and hypoxemic respiratory failure, often requiring mechanical ventilation.34,35 In addition to this clinical presentation, diagnostic criteria include diffuse alveolar or interstitial radiological opacities on imaging, eosinophilia >25% on BAL, exclusion of infectious or other causes of pulmonary eosinophilia, and rapid response to treatment with corticosteroids.41 It is not usually accompanied by peripheral eosinophilia.34,35,38,42 In some cases, it has resolved spontaneously.40 Cases of AEP recurrence have also been reported after the resumption of smoking.39,43
On HRCT, it manifests as diffuse mixed, consolidative alveolar, or ground-glass pulmonary opacities associated with an interstitial septal pattern. The abnormalities generally predominate in the lung bases and association with unilateral or bilateral pleural effusion is common11 (Fig. 5).
 and coronal reconstruction (image d) revealing extensive bilateral alveolar opacities (blue arrows) associated with septal thickening, mainly at the bases, creating a crazy-paving pattern. Bilateral pleural effusion.")
AEP. An 18-year-old woman admitted to the ICU for acute respiratory failure with AEP diagnostic criteria, in addition to peripheral eosinophilia >15%. The clinical and radiological picture resolves rapidly in two to three days after the administration of boluses of corticosteroids. Axial HRCT slices (images a–c) and coronal reconstruction (image d) revealing extensive bilateral alveolar opacities (blue arrows) associated with septal thickening, mainly at the bases, creating a crazy-paving pattern. Bilateral pleural effusion.
The histopathological picture corresponds to diffuse alveolar damage associated with interstitial and alveolar infiltrates of eosinophils, representing the distinguishing feature11 (Fig. 6).
 (H&E 2⋅, 5⋅ and 10⋅).")
Although biopsy is not necessary in many cases if the diagnostic criteria are met, the clinical picture is not specific and may overlap with other causes of acute respiratory failure. Radiologically, AEP may be indistinguishable from bilateral severe infectious pneumonias, other causes of diffuse alveolar damage (DAD), alveolar haemorrhage, and pulmonary oedema.
Pulmonary fibrosis and smokingSince the first description of fibrosis in the alveolar wall in the lungs of smokers,44,45 the association between tobacco smoke and the development of pulmonary fibrotic changes other than IPF, both in terms of clinical and histological features, has been extensively documented.11,12,30,44,46–49 The extent of these changes has even been related to the level of accumulated tobacco consumption.44
Emphysema very frequently coexists with all smoking-related interstitial lung abnormalities. Some also report a higher prevalence of emphysema in smokers with the interstitial disease.50 Although the definition of emphysema excludes evident fibrosis,51 the presence of microscopic fibrosis in CLE is widespread, even more so in paraseptal emphysema (PSE), which is why some consider fibrosis to be part of the emphysematous process.10
The interpretation of the radiological patterns in the case of smoking-related interstitial disease with fibrosis entails an added difficulty that comes from both the coexistence with pulmonary emphysema and the overlap that occurs with the other fibrosing histological patterns.2,9,12,52 From the radiological point of view, it is important to differentiate the patterns of fibrotic UIP and NSIP from the other patterns associated with smoking, as they have a worse prognosis. Specifically, the main difficulty lies in differentiating between honeycomb cysts and emphysema.
Smoking-related interstitial fibrosisSRIF is an entity described by Katzenstein7 and Katzenstein et al.,7,49 which refers to a pattern of interstitial fibrosis associated with emphysema with well-differentiated histological characteristics, frequently present in smokers. It has been termed differently according to the authors: SRIF (smoking-related interstitial fibrosis),7 RB-ILD (respiratory bronchiolitis-associated interstitial lung disease) with fibrosis53,54 or AEF (air space enlargement with fibrosis).55 Its prevalence varies, depending on the series, between 14% and 60%.49,53,55
A thickening of the alveolar septa characterises the unique histopathological picture that defines this entity due to a uniform, dense deposit of thick bundles of eosinophilic collagen with a hyaline appearance that may be interspersed with bands of hypertrophic smooth muscle.7 It is not particularly cellular, with minimal associated inflammation. It adopts a peribronchiolar and subpleural distribution. CLE and RB are almost always present, and the lung architecture is preserved, with distortion in the form of a scar or honeycombing very rare.49 Because it is a well-defined clinical picture, it can be distinguished with considerable certainty from other forms of interstitial lung disease.9 (Figs. 7 and 8).
 reveal mild subpleural reticulation and centrilobular nodularity, with some small peripheral cysts on the right base (image d), associated with mild ground-glass opacities (blue arrows). In addition, signs of bronchopathy secondary to smoking are observed. Histological section (image f): subpleural interstitial fibrosis (orange arrow) due to thickened, collagenised alveolar septa without inflammatory infiltrate. Alveolar spaces with discrete emphysema and intraalveolar macrophages (shorter orange arrow) (H&E 10⋅).")
SRIF in a 66-year-old man, heavy smoker. Axial HRCT slices (images a–e) reveal mild subpleural reticulation and centrilobular nodularity, with some small peripheral cysts on the right base (image d), associated with mild ground-glass opacities (blue arrows). In addition, signs of bronchopathy secondary to smoking are observed. Histological section (image f): subpleural interstitial fibrosis (orange arrow) due to thickened, collagenised alveolar septa without inflammatory infiltrate. Alveolar spaces with discrete emphysema and intraalveolar macrophages (shorter orange arrow) (H&E 10⋅).
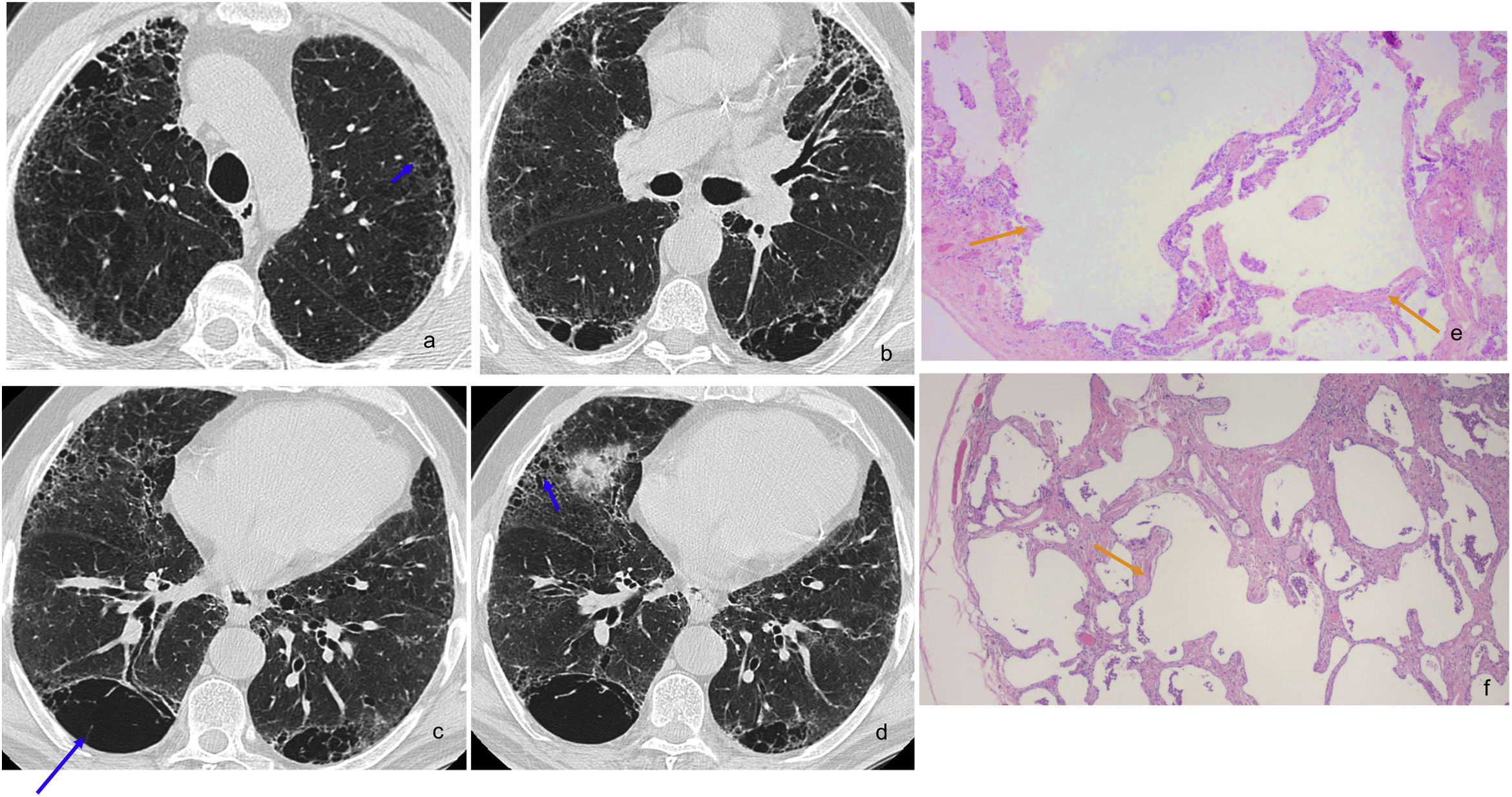
 showing large CLE and PSE cystic spaces with walls, some septated due to probable confluence (long blue arrow). In addition, there are numerous small peripheral cysts with thin walls (short blue arrows) that could mimic honeycombing; however, many of them are in the upper lobes, more or less separated from the pleural surface. Histological sections: image e, with subpleural emphysema (orange arrows) (H&E 5⋅) and image f, with findings of subpleural interstitial fibrosis due to thickened, collagenised alveolar septa without inflammatory infiltrate; alveolar spaces with mild emphysema (orange arrow) and intraalveolar macrophages (H&E 10⋅).")
SRIF in a 67-year-old man, a heavy smoker, almost asymptomatic, with an obstructive spirometry pattern and a slight decrease in diffusion, which did not progress over several years. Axial HRCT slices (images a–d) showing large CLE and PSE cystic spaces with walls, some septated due to probable confluence (long blue arrow). In addition, there are numerous small peripheral cysts with thin walls (short blue arrows) that could mimic honeycombing; however, many of them are in the upper lobes, more or less separated from the pleural surface. Histological sections: image e, with subpleural emphysema (orange arrows) (H&E 5⋅) and image f, with findings of subpleural interstitial fibrosis due to thickened, collagenised alveolar septa without inflammatory infiltrate; alveolar spaces with mild emphysema (orange arrow) and intraalveolar macrophages (H&E 10⋅).
For some authors, SRIF does not have defined radiological characteristics49; for others, the pattern is similar to the RB-ILD pattern, with micronodularity and ground-glass opacities53 (Fig. 7); for others, there is mild reticulation and ground-glass opacities associated with emphysema in the upper lung fields54; “honeycombing” has even been described with the subpleural lung preserved and adjacent areas of emphysema.52 The latter coincides with the experience of Iwasawa et al.,12 who found variable patterns in HRCT in patients with a histological diagnosis of SRIF, which included thin-walled air cysts or small cysts in the areas of reticulation, in some cases very difficult to differentiate from the UIP pattern. This apparent radiological image of honeycombing can be explained by the fibrosis's location around emphysema spaces in the subpleural parenchyma.8 SRIF cysts have thin walls and a preference for the upper lobes and middle areas of the lower lobes, slightly separated from the pleural surface, unlike the honeycombing of UIP56 (Fig. 8).
In most patients, this SRIF is an incidental finding in lung tissue biopsied for other reasons, not necessarily with radiological translation. These patients do not usually present with symptoms; when they do, they are non-specific, with cough and dyspnoea being the most common. Pulmonary function tests (PFTs) may show a mild or moderate restrictive pattern, along with decreased diffusing capacity for carbon monoxide (DLCO). The clinical course of SRIF is favourable, generally stable or with minimal progression.7,49,53
Unlike the other diseases in this review recognised as smoking-related clinical entities, each with a specific histopathological substrate, SRIF has been considered an entity whose diagnosis is based exclusively on a singular histological picture.1,7–9,12,16,24,49,53 Its importance may lie in the fact that, despite being an incidental condition without clinical significance, it is widespread in smokers, in whom it occurs in isolation or coexisting with other interstitial diseases associated with smoking, or idiopathic, that in fact, may be clinically relevant, such as UIP or fibrotic NSIP. It involves abnormalities in HRCT that can overlap and be difficult to differentiate from those associated with these other entities and can even mimic honeycombing. It is, therefore, essential to establish the clinical and radiological differential diagnosis with these entities in the presence of symptoms of progressive exertional dyspnoea and cough associated with significant functional limitation and DLCO. In these cases, a biopsy may be essential.8,9
Non-specific interstitial pneumoniaLung damage expressed with the histological and radiological pattern of NSIP may have many different aetiologies, including systemic autoimmune disease, HP, and drug-induced lung disease.57–59 The histological pattern is characterised by the uniform occupation of the alveolar interstitium by fibrosis and inflammation of varying proportions, on which the classification into “cellular” or “fibrotic” depends.57,60 Foci of fibroblasts may appear less frequently than in UIP, and areas of organising pneumonia superimposed on chronic interstitial changes.57 Classic findings on HRCT include symmetrical ground-glass opacities with underlying reticulation and traction bronchiolectasis, with little or no honeycombing.32,58,59
Some authors propose a pathogenic association between smoking and a pattern of fibrotic NSIP.21,50,53,61,62 They even speculate that the fibrosis that follows the NSIP pattern in smokers could represent a fibrotic stage of DIP in which the macrophages have been cleared from the alveoli.6,11,21 One of these researchers found a higher prevalence of emphysema in smokers with a fibrotic NSIP pattern than in smokers without interstitial abnormalities.50
Iwasawa et al.12 state that it is very difficult to differentiate in the initial HRCT between fibrotic NSIP, DIP or the coexistence of both patterns, which is common. However, this distinction is relevant since the progression of DIP to severe fibrosis is unlikely, in which case we would be facing a pattern of NSIP with or without DIP overlap.29
The overlapping of the NSIP and DIP patterns is also recognised, and it is postulated that it is more common in smokers.2,52 In a lung cancer early detection programme with CT, 37% of the cases in which interstitial abnormalities with a fibrotic pattern were identified had a mixed pattern with UIP and NSIP findings.2
Idiopathic pulmonary fibrosis/usual interstitial pneumoniaIPF is a chronic idiopathic and fibrosing interstitial disease with a poor prognosis, with a pattern of UIP as histopathological substrate, which has its radiological correlate. The UIP pattern has been defined by the Fleishner society, which has established subpleural involvement predominantly at baseline, with heterogeneous distribution and honeycombing, with or without bronchiectasis and traction bronchiolectasis as radiological diagnostic criteria.63 Smoking is considered a probable risk factor for the development of IPF. In addition, the survival of smokers with IPF is lower than of non-smokers.63,64
The histological pattern in smokers has a greater tendency to overlap with histological features of fibrotic NSIP2,52 as mentioned in the previous section. In addition, other smoking-related idiopathic ILD patterns may overlap and often coexist with emphysema. All this can complicate the interpretation of both the histological and radiological pattern.
Honeycombing is a characteristic diagnostic feature of UIP, both histologically and on HRCT. Honeycomb cysts and CLE coexist in the same lobe in smokers with IPF,10 which poses many difficulties for the identification of and differentiation between findings.10,12 Fibrosis in UIP is located on the periphery of the lung lobule and is characterised by alveolar collapse65 and volume loss, while emphysema is centrilobular. Cystic images with central points, representing remnants of distal vessels and bronchi, may help differentiate emphysema on HRCT.12 On the other hand, honeycomb cysts in the histological study usually measure less than 1mm and are often filled with mucus,66 so they may not be seen on HRCT. Some authors maintain that HRCT honeycombing correlates better with the histological substrate of bronchiolectasis.67
It is concluded from all this that the recognition of the honeycomb pattern is especially complicated in smokers with fibrosing ILD.
Combined pulmonary fibrosis and emphysemaCombined pulmonary fibrosis and emphysema (CPFE) denominate an entity initially described by Cottin et al.68 which is increasingly recognised and which not only refers to the superimposition of emphysema with pulmonary fibrosis but is thought to represent a clinical syndrome with distinct features.69 It is more common in male smokers older than 65,68 with cumulative consumption greater than 40 pack-years,70 generally very symptomatic, with cough and dyspnoea accompanied by severe exertional hypoxemia, and very low carbon monoxide diffusion coefficients, in contrast to preserved spirometric values.68,70 Almost all the authors report a high prevalence of PHTN in these patients, at the time of diagnosis or during the evolution, which ranges between 50% and 90% of cases,68,71,72 and is greater than in IPF not associated with emphysema.71,73 This association entails a worse prognosis,72 with a one-year survival of 60% in some series.71. Although initial studies reported a median survival of six years,68 greater than that of IPF, other more recent ones report survivals of fewer than five years and higher mortality than IPF.74 This may be due to the higher incidence of lung cancer associated with CPFE in relation to IPF.71,73,75
HRCT findings are characterised by the presence of CLE and PSE in the upper lung fields and predominantly basal fibrosis (Fig. 9a–f). Some authors believe that a minimum extension of emphysema of at least 10% of the lung volume is necessary.76 Wide cystic spaces with fibrous walls may appear, both in the upper and lower lobes, which represent SRIF changes for some authors.10,55,77,78 In addition to emphysematous changes, basal fibrosis often presents with reticulation, honeycombing, architectural distortion, and bronchiolectasis.68,68,78 However, in some series, ground-glass opacities predominate with a pattern more indicative of fibrotic NSIP.68
 reveal large emphysematous bullae (blue arrows) in the upper lobes and coarse reticular opacities with predominantly basal bronchiectasis and traction bronchiolectasis, without honeycombing (orange arrows). Histological slices (g–h) with an indeterminate fibrosing pattern. Image g: shows subpleural and bronchiolocentric fibrosis, with architectural distortion, emphysema, and bronchiolectasis (H&E 5⋅); image h: with fibrosis and emphysema with pronounced architectural distortion (H&E 5⋅).")
CPFE in a man with accumulated tobacco consumption of 35 pack-years, with an indeterminate fibrosing histological pattern in the lung biopsy. HRCT (images a–f) reveal large emphysematous bullae (blue arrows) in the upper lobes and coarse reticular opacities with predominantly basal bronchiectasis and traction bronchiolectasis, without honeycombing (orange arrows). Histological slices (g–h) with an indeterminate fibrosing pattern. Image g: shows subpleural and bronchiolocentric fibrosis, with architectural distortion, emphysema, and bronchiolectasis (H&E 5⋅); image h: with fibrosis and emphysema with pronounced architectural distortion (H&E 5⋅).
The histological pattern of fibrosis most commonly associated with CPFE is the UIP pattern, although the fibrotic NSIP pattern has also been described.11,12,69,79 Patterns of fibrosis in CPFE are generally very heterogeneous from the histological point of view, due to overlapping lesions attributable to smoking, frequently including SRIF, which can be associated with thick-walled hyalinised subpleural cysts. However, SRIF has not been established as a cause of CPFE.69 Therefore, differentiation between fibrosing UIP or NSIP and SRIF is essential. The difficulty in classifying the histological pattern is reflected in the interpretation of the radiological pattern. The clinical importance of this differentiation from the prognostic point of view remains to be defined.
ConclusionsThe spectrum of smoking-related ILD encompasses a heterogeneous group of diseases that, although described and classified as individualised entities, tend to coexist and present overlapping histological patterns in the same patient, which is also reflected in radiological findings on HRCT and poses significant diagnostic challenges.
SRIF is a defined histopathological picture frequently present in lung biopsies from smokers associated with emphysema. It may be reflected in HRCT findings, especially when it mimics honeycombing, which poses difficulties in the differential diagnosis with clinically relevant entities such as UIP and CPFE.
The key to the diagnosis is to discern which patterns may be responsible for the clinical picture and have a greater prognostic implication.
From all of the above, it can be deduced that a multidisciplinary approach, from a joint histopathological, clinical and radiological perspective, is essential in smoking-related ILD.
Authorship- 1.
Responsible for the integrity of the study: PSG, SNH, LGI and JARP.
- 2.
Study conception: PSG, SNH, LGI and JARP.
- 3.
Study design: PSG, SNH and LGI.
- 4.
Data collection: N/A.
- 5.
Data analysis and interpretation: N/A.
- 6.
Statistical processing: N/A.
- 7.
Literature search: PSG and SNH.
- 8.
Drafting of the article: PSG, SNH and LGI.
- 9.
Critical review of the manuscript with intellectually significant contributions: PSG, SNH, LGI and JARP.
- 10.
Approval of the final version: PSG, SNH, LGI and JARP.
The authors declare that they have no conflicts of interest.

