




Bacillus thuringiensis is an entomopathogen belonging to the Bacillus cereus clade. We isolated a tetracycline-resistant strain called m401, recovered it from honey, and identified it as Bacillus thuringiensis sv. kumamotoensis based on the average nucleotide identity calculations (ANIb) comparison and the analysis of the gyrB gene sequences of different B. thuringiensis serovars. Sequences with homology to virulence factors [cytK, nheA, nheB, nheC, hblA, hblB, hblC, hblD, entFM, and inhA] and tetracycline resistance genes [tet(45), tet(V), and tet(M)/tet(W)/tet(O)/tet(S) family] were identified in the bacterial chromosome. The prediction of plasmid-coding regions revealed homolog sequences to the MarR and TetR/AcrR family of transcriptional regulators, toxins, and lantipeptides. The genome mining analysis revealed 12 regions of biosynthetic gene clusters responsible for synthesizing secondary metabolites. We identified biosynthetic gene clusters coding for bacteriocins, siderophores, ribosomally synthesized post-translationally modified peptide products, and non-ribosomal peptide synthetase clusters that provide evidence for the possible use of Bt m401 as a biocontrol agent. Furthermore, Bt m401 showed high inhibition against all Paenibacillus larvae genotypes tested in vitro. In conclusion, Bt m401 owns various genes involved in different biological processes, such as transductional regulators associated with antibiotic resistance, toxins, and antimicrobial peptides with potential biotechnological and biocontrol applications.
Bacillus thuringiensis es un microorganismo entomopatógeno perteneciente al clado Bacillus cereus. En este trabajo, aislamos de miel una cepa resistente a tetraciclina y la identificamos como Bacillus thuringiensis sv. kumamotoensis (Bt m401) mediante comparación de genomas completos y cálculo de identidad promedio de nucleótidos (ANIb), utilizando la base de datos EDGAR. Este resultado fue corroborado por el análisis filogenético del gen gyrB, que permite diferenciar serovares de B. thuringiensis. Se confirmó la presencia del gen de resistencia a tetraciclina tet(45) en el cromosoma bacteriano y se identificaron secuencias homólogas a genes de virulencia (cytK, nheA, nheB, nheC, hblA, hblB, hblC, hblD y entFM). El mobiloma de Bt m401 está constituido por 4 plásmidos y se identificaron secuencias homólogas a reguladores transduccionales (MarR y TetR/AcR), a toxinas (Cry1 y zeta toxin) y a péptidos antimicrobianos (mersacidin family lantibiotics). La cepa Bt m401 demostró una muy buena actividad antagónica in vitro contra distintos genotipos de Paenibacillus larvae, agente causal de la loque americana de las abejas. El estudio de minería genómica para grupos de genes biosintéticos reveló 12 regiones de clústeres de genes responsables de la síntesis de metabolitos secundarios. Se identificaron clústeres que codifican bacteriocinas, sideróforos, RiPP-like y NRPS y lantipéptidos. En conclusión, Bt m401 presentó una amplia variedad de genes que estarían involucrados en diferentes procesos biológicos, con un potencial interesante para su empleo en aplicaciones biotecnológicas.
Bacillus thuringiensis (Bt) is a ubiquitous Gram positive, spore-forming bacterium isolated worldwide from a great diversity of sources, such as soil, water, insects, dust, phyllosphere, and diverse foods20,37. Bt belongs to the Cereus clade consisting of 17 species, including Bacillus anthracis, Bacillus cereus, Bacillus mycoides, Bacillus thuringiensis, and Bacillus toyonensis, among others26.
Bt is the most successful microbial insecticide against different insect pest orders in agriculture35,37. Its action is based on insecticidal toxins that are active during the pathogenic process by synthesizing parasporal crystalline inclusions containing Cry and Cyt proteins. Some Bt strains also synthesize insecticidal proteins during the vegetative growth phase, known as vegetative insecticidal proteins (Vips), and secreted insecticidal proteins (Sips) that hold insecticidal activity against lepidopteran, coleopteran, and some homopteran pests14,37.
Bt-related studies mainly focused on insecticidal activities; nevertheless, it is also considered a plant growth-promoting bacterium (PGPB)11,25. Moreover, many Bt strains produce virulence factors and other metabolites, including a broad range of antimicrobial peptides that are active against pathogenic bacteria and fungi, e.g., cyclic lipopeptides, lanthipeptides, polyketides, bacteriocins, and bacteriocin-like inhibitors (BLIS)2,11,25,30,36,40,43. Diverse antimicrobial peptides have been isolated from different Bt strains showing wide or narrow bactericidal or bacteriostatic effects. Within this group, bacthuricins, entomocins, fengycins, thuricins, thurincin H, and thumolicin, among others, were reported2,22,25,29,30,36,49.
American foulbrood (AFB) is the most widespread and destructive disease of bacterial origin that affects the larval and pupal stages of honeybees (Apis mellifera L.)24. In North and South American honey-producing countries, beekeepers have routinely treated colonies with oxytetracycline (OTC) to control AFB38. The widespread use of OTC has increased tetracycline resistance (TETR) in Paenibacillus larvae populations by enhancing the transfer of TETR-encoding plasmids containing tet(L) genes4. The tetL determinant found in P. larvae is nearly identical, in sequence (99–100% identity), to one of the resistance loci harbored by honeybee gut symbionts, suggesting past horizontal transfer between commensal gut bacteria and this pathogen46.
Spore-forming bacteria of the genus Bacillus, including Bt, are frequently present in honey and other apiarian sources1,7 and can act as reservoirs and environmental suppliers of antibiotic resistance enabling the spread of TETR genes between different bacterial species46. Bacillus species are also promising sources of bioactive compounds and secondary metabolites with potential uses beyond the field of apiculture, including a broad range of antimicrobial compounds with activity against pathogenic bacteria and fungi2,12,22,29,36,43.
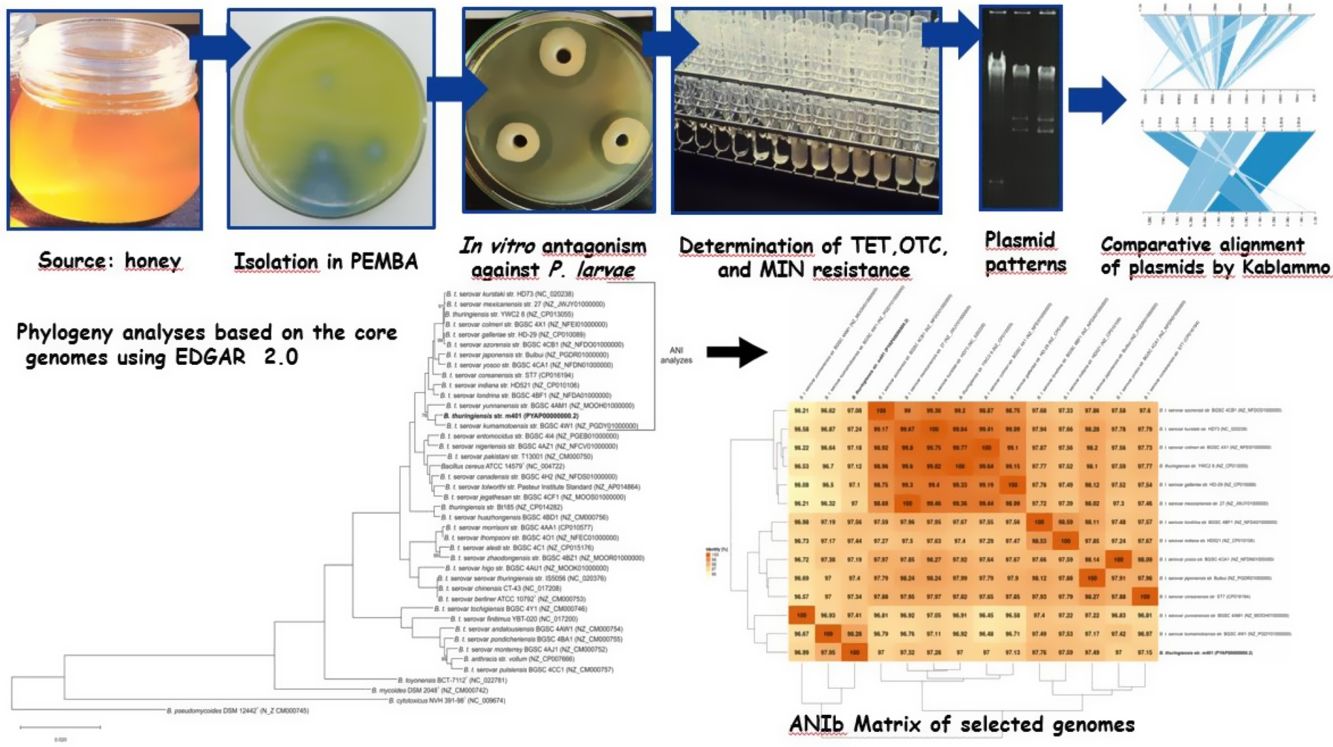
In previous studies, we evaluated several Bacillus strains for their in vitro antimicrobial activity against P. larvae, the causal agent of AFB6,12,34. These inhibitory effects correlated with BLIS production34 and other antimicrobial peptides12. The present paper presents further insight into the genome of Bacillus thuringiensis strain m401 isolated from honey. We performed a comparative analysis with other Bt strains mainly focused on detecting genes encoding antibiotic resistance, antimicrobial peptides, toxins, and related regulatory gene factors. At the same time, we investigated the in vitro antagonism of Bt m401 against 52 strains of P. larvae belonging to different genotypes from diverse geographical areas and with different tetracycline sensitivity.
Materials and methodsBacterial strains and culture conditionsBt m401 was isolated from a honey sample from Pigüe, Argentina (37°36′S; 64°24′W), using polymyxin-pyruvate-egg-yolk-mannitol agar (PEMBA) as previously described31. The isolate was kept as frozen stocks at −80°C in trypticase soy broth (TSB) containing 20% v/v glycerol. The P. larvae strains (n=52) used were obtained from different geographical areas (Table 1). The collection had been previously characterized by rep-PCR with primers BOX and ERIC6 and tetracycline resistance4,5. P. larvae isolates were preserved in Müeller Hinton broth-yeast extract-potassium phosphate-glucose-pyruvate (MYPGP) broth21 containing 20% v/v glycerol. For short-term storage, the strains were kept at 4°C in the appropriate semi-solid medium.
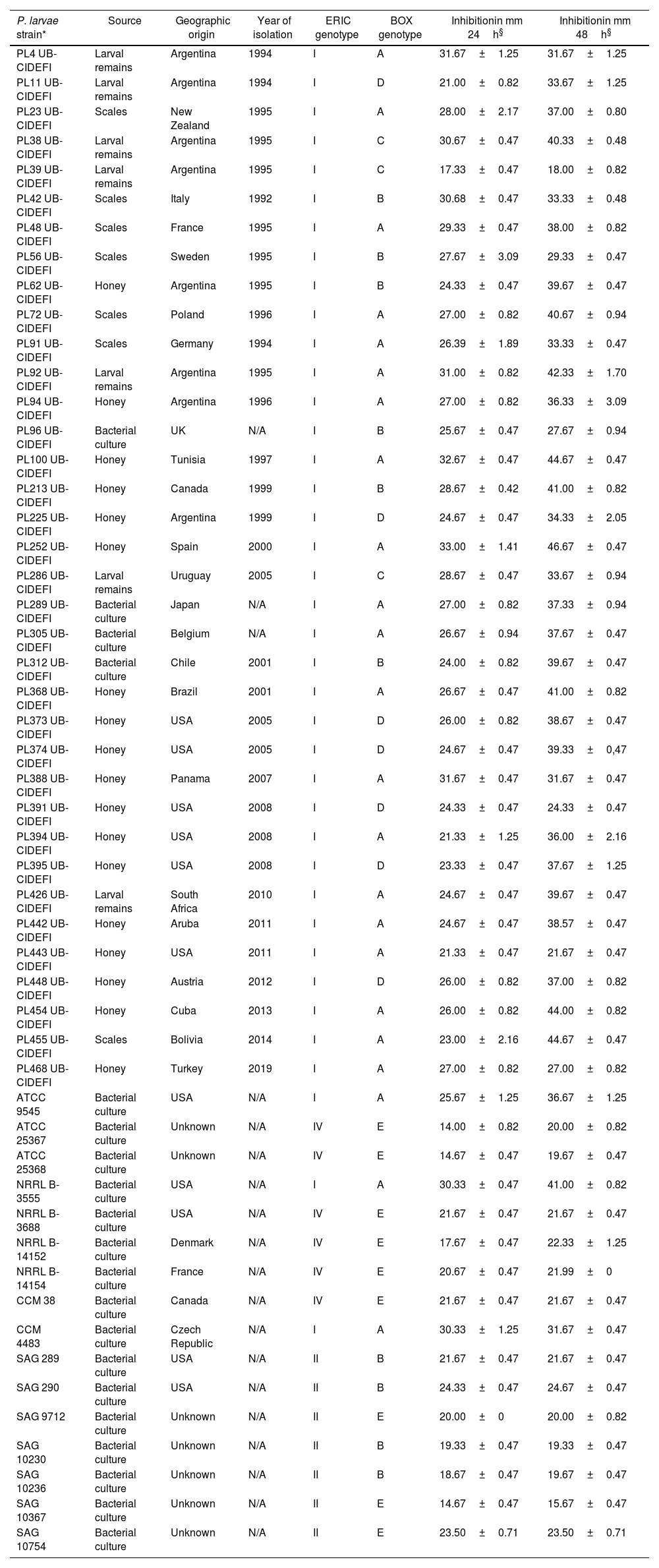
Antibacterial spectrum of Bt m401 against Paenibacillus larvae strains as determined by the well diffusion technique
| P. larvae strain* | Source | Geographic origin | Year of isolation | ERIC genotype | BOX genotype | Inhibitionin mm 24h§ | Inhibitionin mm 48h§ |
|---|---|---|---|---|---|---|---|
| PL4 UB-CIDEFI | Larval remains | Argentina | 1994 | I | A | 31.67±1.25 | 31.67±1.25 |
| PL11 UB-CIDEFI | Larval remains | Argentina | 1994 | I | D | 21.00±0.82 | 33.67±1.25 |
| PL23 UB-CIDEFI | Scales | New Zealand | 1995 | I | A | 28.00±2.17 | 37.00±0.80 |
| PL38 UB-CIDEFI | Larval remains | Argentina | 1995 | I | C | 30.67±0.47 | 40.33±0.48 |
| PL39 UB-CIDEFI | Larval remains | Argentina | 1995 | I | C | 17.33±0.47 | 18.00±0.82 |
| PL42 UB-CIDEFI | Scales | Italy | 1992 | I | B | 30.68±0.47 | 33.33±0.48 |
| PL48 UB-CIDEFI | Scales | France | 1995 | I | A | 29.33±0.47 | 38.00±0.82 |
| PL56 UB-CIDEFI | Scales | Sweden | 1995 | I | B | 27.67±3.09 | 29.33±0.47 |
| PL62 UB-CIDEFI | Honey | Argentina | 1995 | I | B | 24.33±0.47 | 39.67±0.47 |
| PL72 UB-CIDEFI | Scales | Poland | 1996 | I | A | 27.00±0.82 | 40.67±0.94 |
| PL91 UB-CIDEFI | Scales | Germany | 1994 | I | A | 26.39±1.89 | 33.33±0.47 |
| PL92 UB-CIDEFI | Larval remains | Argentina | 1995 | I | A | 31.00±0.82 | 42.33±1.70 |
| PL94 UB-CIDEFI | Honey | Argentina | 1996 | I | A | 27.00±0.82 | 36.33±3.09 |
| PL96 UB-CIDEFI | Bacterial culture | UK | N/A | I | B | 25.67±0.47 | 27.67±0.94 |
| PL100 UB-CIDEFI | Honey | Tunisia | 1997 | I | A | 32.67±0.47 | 44.67±0.47 |
| PL213 UB-CIDEFI | Honey | Canada | 1999 | I | B | 28.67±0.42 | 41.00±0.82 |
| PL225 UB-CIDEFI | Honey | Argentina | 1999 | I | D | 24.67±0.47 | 34.33±2.05 |
| PL252 UB-CIDEFI | Honey | Spain | 2000 | I | A | 33.00±1.41 | 46.67±0.47 |
| PL286 UB-CIDEFI | Larval remains | Uruguay | 2005 | I | C | 28.67±0.47 | 33.67±0.94 |
| PL289 UB-CIDEFI | Bacterial culture | Japan | N/A | I | A | 27.00±0.82 | 37.33±0.94 |
| PL305 UB-CIDEFI | Bacterial culture | Belgium | N/A | I | A | 26.67±0.94 | 37.67±0.47 |
| PL312 UB-CIDEFI | Bacterial culture | Chile | 2001 | I | B | 24.00±0.82 | 39.67±0.47 |
| PL368 UB-CIDEFI | Honey | Brazil | 2001 | I | A | 26.67±0.47 | 41.00±0.82 |
| PL373 UB-CIDEFI | Honey | USA | 2005 | I | D | 26.00±0.82 | 38.67±0.47 |
| PL374 UB-CIDEFI | Honey | USA | 2005 | I | D | 24.67±0.47 | 39.33±0,47 |
| PL388 UB-CIDEFI | Honey | Panama | 2007 | I | A | 31.67±0.47 | 31.67±0.47 |
| PL391 UB-CIDEFI | Honey | USA | 2008 | I | D | 24.33±0.47 | 24.33±0.47 |
| PL394 UB-CIDEFI | Honey | USA | 2008 | I | A | 21.33±1.25 | 36.00±2.16 |
| PL395 UB-CIDEFI | Honey | USA | 2008 | I | D | 23.33±0.47 | 37.67±1.25 |
| PL426 UB-CIDEFI | Larval remains | South Africa | 2010 | I | A | 24.67±0.47 | 39.67±0.47 |
| PL442 UB-CIDEFI | Honey | Aruba | 2011 | I | A | 24.67±0.47 | 38.57±0.47 |
| PL443 UB-CIDEFI | Honey | USA | 2011 | I | A | 21.33±0.47 | 21.67±0.47 |
| PL448 UB-CIDEFI | Honey | Austria | 2012 | I | D | 26.00±0.82 | 37.00±0.82 |
| PL454 UB-CIDEFI | Honey | Cuba | 2013 | I | A | 26.00±0.82 | 44.00±0.82 |
| PL455 UB-CIDEFI | Scales | Bolivia | 2014 | I | A | 23.00±2.16 | 44.67±0.47 |
| PL468 UB-CIDEFI | Honey | Turkey | 2019 | I | A | 27.00±0.82 | 27.00±0.82 |
| ATCC 9545 | Bacterial culture | USA | N/A | I | A | 25.67±1.25 | 36.67±1.25 |
| ATCC 25367 | Bacterial culture | Unknown | N/A | IV | E | 14.00±0.82 | 20.00±0.82 |
| ATCC 25368 | Bacterial culture | Unknown | N/A | IV | E | 14.67±0.47 | 19.67±0.47 |
| NRRL B-3555 | Bacterial culture | USA | N/A | I | A | 30.33±0.47 | 41.00±0.82 |
| NRRL B-3688 | Bacterial culture | USA | N/A | IV | E | 21.67±0.47 | 21.67±0.47 |
| NRRL B-14152 | Bacterial culture | Denmark | N/A | IV | E | 17.67±0.47 | 22.33±1.25 |
| NRRL B-14154 | Bacterial culture | France | N/A | IV | E | 20.67±0.47 | 21.99±0 |
| CCM 38 | Bacterial culture | Canada | N/A | IV | E | 21.67±0.47 | 21.67±0.47 |
| CCM 4483 | Bacterial culture | Czech Republic | N/A | I | A | 30.33±1.25 | 31.67±0.47 |
| SAG 289 | Bacterial culture | USA | N/A | II | B | 21.67±0.47 | 21.67±0.47 |
| SAG 290 | Bacterial culture | USA | N/A | II | B | 24.33±0.47 | 24.67±0.47 |
| SAG 9712 | Bacterial culture | Unknown | N/A | II | E | 20.00±0 | 20.00±0.82 |
| SAG 10230 | Bacterial culture | Unknown | N/A | II | B | 19.33±0.47 | 19.33±0.47 |
| SAG 10236 | Bacterial culture | Unknown | N/A | II | B | 18.67±0.47 | 19.67±0.47 |
| SAG 10367 | Bacterial culture | Unknown | N/A | II | E | 14.67±0.47 | 15.67±0.47 |
| SAG 10754 | Bacterial culture | Unknown | N/A | II | E | 23.50±0.71 | 23.50±0.71 |
UB-CIDEFI: Unidad de Bacteriología-Centro de Investigaciones de Fitopatología, Facultad de Ciencias Agrarias y Forestales, Universidad Nacional de La Plata, La Plata, Argentina; ATCC: American Type Culture Collection, Rockville, Maryland, USA; CCM: Czech Collection of Microorganisms, Brno, Czech Republic; NRRL: Northern Utilization Research and Development Division, Peoria, Illinois, USA; SAG: Servicio Agrícola Ganadero, Chile.
To identify the isolate m401 at the species level, bacterial smears were examined for the presence and location of spores within cells and the size and shape of vegetative cells3,31. Unstained globules in the cytoplasm were examined by phase contrast microscopy, and parasporal crystals were single-stained with Coomassie Blue. Colony characteristics and media appearance in Bacillus Chromoselect agar (Sigma-Aldrich®) and PEMBA (Britania®) were evaluated as previously described3,31. Furthermore, the isolate was tested for catalase, production of lecithinase, Voges-Proskauer reaction, mannitol, and arabinose utilization, anaerobic utilization of glucose, hemolytic activity, and starch and gelatin hydrolysis according to standard protocols3,31. Additionally, the identity was confirmed by a PCR-RFLP assay, as described by López and Alippi32.
Genome re-assembly of Bt m401 and confirmation of a new plasmidThe previously assembled genomic DNA of Bt m4011 was reassembled using Unicycler v0.4.7. Bioinformatic evidence showed that scaffold 26 (18160bp) of the old assembly matches the scaffold of the new assembly (19094bp) but with a different relative position. Moreover, Unicycler predicts the new scaffold as a circular replicon, a new plasmid. To confirm this, we designed primers to target the outer region's ends of scaffold 26. Thus, if this amplifies the region, it would confirm the plasmid. The primers Scaff26_F/R were designed using Primer-Blast (www.ncbi.nlm.nih.gov/tools/primer-blast/). Primers Scaff26_F/R sequences were 5′-AGCAAAGTCGTGTATCCTGC-3′ and 5′-TGGTGCGCTTCTAAATGGTG-3′, respectively. The reaction mixture (25μl) contained 2.5μl buffer 1× (Promega®), 0.5mM Scaff26_F/R primers; 1.25mM MgCl2 (Promega®); 1.76mM of each dNTP (Promega®); 1 U T-plus Taq DNA polymerase (Inbio Highway®) and 2μl of DNA (25μg/ml) obtained as described above. The cycling program consisted of a 95°C (5min) step; 35 cycles of 95°C (30s), 58°C for annealing (2min), 72°C (2min); and a final step of 72°C (4min). Amplifications were performed in a thermal cycler Eppendorf Mastercycler (Eppendorf AG, Hamburg, Germany).
PCR amplicons obtained by primers Scaff26_F/R were further confirmed by digestion using restriction enzymes. After amplifying a PCR product of 1917bp, subsamples of 4μl were incubated with endonucleases HaeIII and CfoI according to the manufacturer's specifications (Promega®, Buenos Aires, Argentina). In each case, the expected sizes of the digested fragments were visualized. Plasmid DNA corresponding to scaffold 26 was named pBTm401d. Amplification products were separated in 1.5% agarose gel in 0.5× TBE buffer, stained with ethidium bromide, and visualized with a UV transilluminator (UVP®, Upland, USA). Photographs were digitalized using DigiDoc-It (UVP®, v.1.125, Upland, USA).
Bioinformatic analyses of the genome and prediction of biosynthetic gene clusters in the genomePutative coding regions were determined using GLIMMER v3 and EasyGene, both trained with the B. cereus ATCC 10987 genome (NC_003909). We used the BLASTx program at the NCBI database (www.ncbi.nlm.nih.gov/) for identifying potential protein products, and sequence similarities and identities were analyzed using BLASTn.
Previous reports demonstrated that gyrB gene nucleotide sequence analyses could distinguish Bt serovars44. A phylogenetic tree was constructed using the gyrB nucleotide sequences from Bt m401 and those from Bt serovars aligned using ClustalW. The study involved 59 gyrB sequences of Bt serovars retrieved from GenBank and a sequence of Escherichia coli (NC_000913.3) as an outgroup. The evolutionary history was inferred using the Maximum Likelihood (ML) method based on the General Time Reversible model. A bootstrap resampling analysis employing 1000 pseudoreplicates was used to assess the reliability of the phylogenetic trees, all using the MEGA 7 program.
Moreover, the Bt m401 genome was uploaded to EDGAR Database for comparative analysis. We selected 40 strains of the Cereus clade, including different Bt serovars. A phylogenetic tree on core genome sequences was constructed using EDGAR 2.0. First, EDGAR calculates the core genes of the selected genomes; then, it aligns each core gene and concatenates them13. Afterward, FastTree software (http://www.microbesonline.org/fasttree/) was used to generate maximum-likelihood phylogenetic trees. EDGAR was also used to calculate an average nucleotide identity (ANI) matrix for the selected genomes. The complete genome sequences of 34 Bt serovars and 6 type strains belonging to the Cereus clade were compared, and the ANI matrix for a selected set of the closest genomes (n=13) was made.
Additionally, BLASTn sequences comparisons of Bt m401 plasmids with plasmids of related species were visualized using Kablammo (Kablammo.wasmuthlab.org). GC viewer server (gcviewer download|SourceForge.net) was used to calculate plasmid G+C content.
Biosynthetic gene clusters (BCGs) in the genome and their corresponding secondary metabolites were predicted using the bacterial version of antiSMASH 6.0 (https://antismash.secondarymetabolites.org/). The gene clusters found were cross-referred in the MIBiG repository (https://mibig.secondarymetabolites.org/).
In vitro antagonism against Paenibacillus larvaeThe antibacterial spectrum of Bt m401 against 52 P. larvae strains from diverse geographical origins was determined using a well diffusion technique described by Alippi and Reynaldi6.
Determination of tetracycline, oxytetracycline, and minocycline resistanceMinimal inhibitory concentration (MIC) values of tetracycline (TET) (Sigma®, USA), oxytetracycline (OTC) (Oxoid®, USA), and minocycline (MIN) (Wyeth-Whitehall®, Brazil) were determined by the broth macrodilution method, according to the recommendations of the Clinical and Laboratory Standards Institute17,18. Increasing concentrations between 0.0312 and 256μg/ml TET, OTC, and MIN were tested. Tubes containing Müller-Hinton (MH) broth without antibiotics were used for controls. Vegetative cells of Bt m401 growing on MH agar for 24h at 30±1°C were suspended in sterile distilled water and adjusted to a concentration of 0.5 Mc Farland17,18. Appropriate dilutions of the inoculum were prepared to deliver a final concentration of bacteria of about 5×105CFU/ml. Two replications were tested for each antibiotic concentration. Inoculated tubes were examined after 24–48h incubation at 37±1°C in constant agitation. Staphylococcus aureus ATCC 29213 and Escherichia coli ATCC 25922 were used for quality control.
Visualization of plasmid DNABacterial cells were screened for extrachromosomal DNA using the technique described by Fagundes and co-workers23. Bt m401 was grown in 50ml Spizizen broth23, supplemented with 16μg/ml TET, and incubated at 30±1°C overnight under constant agitation. P. larvae strain PL373, containing two plasmids of approximately 5000bp and 8000 bp4, was used as a control and was grown in MYPGP broth supplemented with 16μg/ml TET and incubated at 37±1°C. Gels were visualized with a UV transilluminator, as described before.
Plasmid and total DNA extractionA rapid procedure using whole cells from plates was employed for total genomic DNA preparations7, while plasmid DNA was prepared by alkaline lysis with the SDS procedure described by Sambrook and Russell41.
PCR analysis of tetracycline-resistant genesThe presence of tet(K), tet(L), tet(M), tet(O), tet(W), otr(A), and otr(B) genes was assessed by PCR as described elsewhere33,45,47. In the case of the tetracycline-resistant tet(45) gene, the primers used were Tet45-F and Tet45-R as described by You and co-workers48 but using 1mM of primers and a final volume of 30μl for the premix. This selection included the most commonly detected tet genes in Bacillus (http://faculty.washington.edu/marilynr/tetweb3.pdf). Both genomic and plasmid DNA were used as templates, and the amplification products were separated and visualized in 1.5% agarose gels in TBE buffer as previously described.
Phylogenetic analysis of tet(45) geneThe tet(45) gene found in Bt m401 and other tet(45) and tet(L) tetracycline-resistant genes retrieved from GenBank were aligned using Clustal W. The analysis involved 14 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 743 positions in the final dataset. The amino acid sequence of the tet(M) gene from Clostridium difficile CD2386 (JN846696.1) was used as an outgroup. The tree was constructed using the ML method based on the General Time Reversible model. A bootstrap resampling analysis employing 1000 pseudoreplicates was used to assess the reliability of the phylogenetic trees, all using MEGA 7.
ResultsSpecies identificationBt m401 stained Gram positive and was facultative anaerobic and catalase-positive. The strain showed ellipsoidal spores not distending the sporangia in a central position. As seen by phase contrast microscopy, the cytoplasm was filled with unstained globules. Moreover, parasporal crystals were detected in stained preparations. Bacterial colonies in Bacillus Chromoselect agar were light blue over a pinkish medium, while in PEMBA, Bt m401 produced crenated colonies of turquoise blue surrounded by egg-yolk precipitation haloes as a result of lecithinase activity. The strain hydrolyzed gelatin and starch and was positive for Voges-Proskauer, and reduction of nitrates to nitrites and negative for mannitol and arabinose utilization. It also showed hemolytic activity in blood agar plates. Additionally, the isolate showed the expected restriction patterns for Bacillus thuringiensis when using a combination of AluI and CfoI enzymes32.
Bioinformatic analyses of the genomePreviously, we sequenced and assembled the Bt m401 genome containing one chromosome and three plasmids1. However, experimental assays suggested the presence of a fourth plasmid (data not shown). In the present work, we reassembled the genome using Unicycler v0.4.7 to uncover potential genes related to tetracycline resistance, virulence factors, and antimicrobial peptides. The final draft genome assembly consists of 45 scaffolds generating a 6005135bp genome size with an average GC content of 34.72% (Table S1). The data set comprises a chromosome and four circular plasmids, which vary from 8307 to 69591bp (Table S2). Plasmids pBTm401a, pBTm401b, and pBTm401c coincided with the first assembly, and a new plasmid of 19094bp, pBTm401d, was detected and assembled. PCR and enzymatic digestions confirmed the circular DNA of pBTm401d (Table S2).
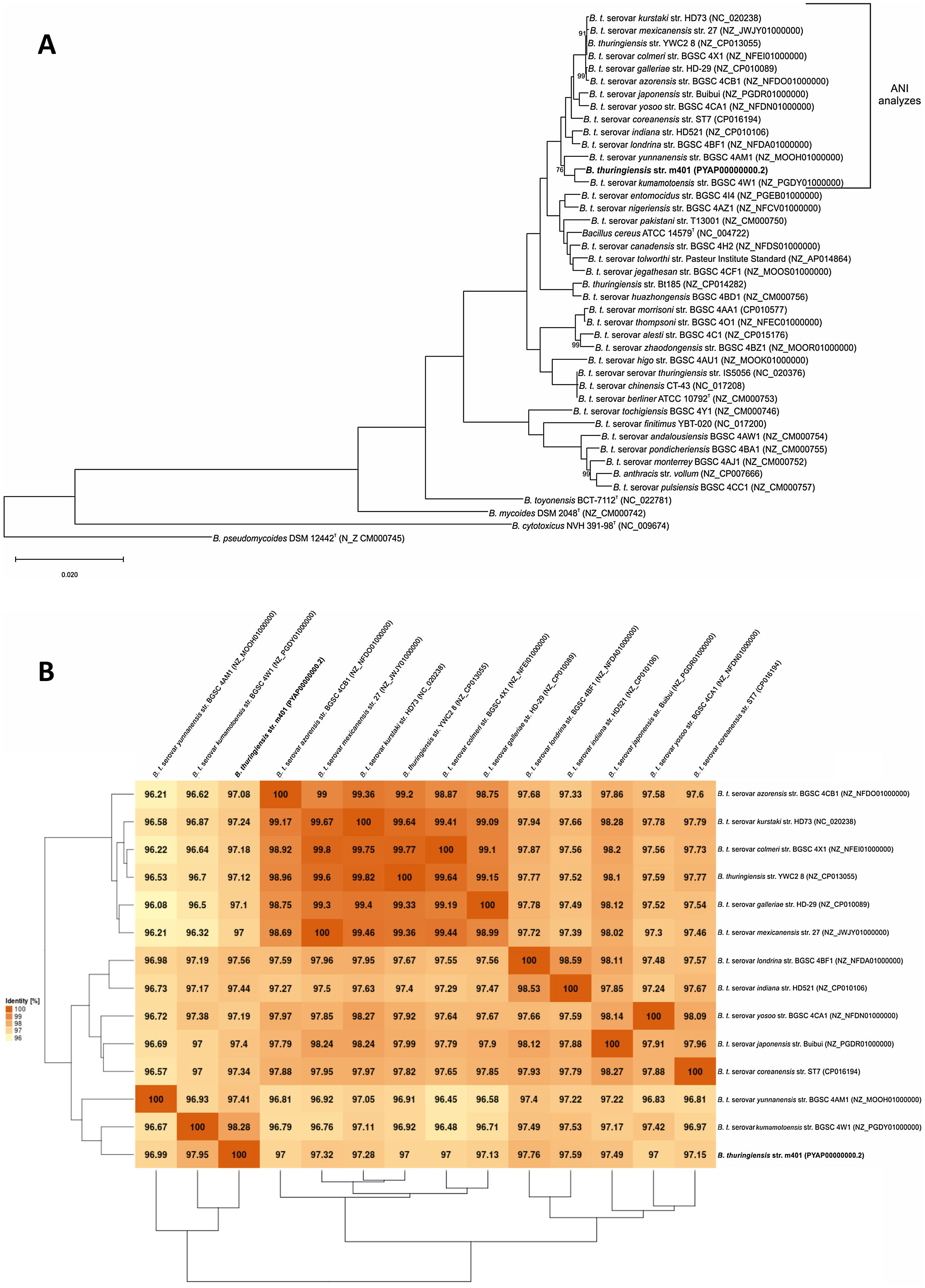
The phylogenetic relationships based on gyrB sequences between Bt m401 and different serovars of Bt are shown in Figure 1. The 52 Bt serovars were separated into 5 clusters (I, II, III, IV, and V). Bt m401 was located in cluster I, revealed at 93% nucleotide sequence identities, together with 21 serovars, including kumamotoensis, yunnanensis, indiana, and londrina, among others.
 are shown next to the branches. All positions containing gaps and missing data were eliminated. E. coli K12 (NC_000913.3) was used as an outgroup. Evolutionary analyses were conducted in MEGA7.")
Bootstrapped Maximum Likelihood tree of different Bacillus thuringiensis serovars generated from the alignment of gyrB nucleotide sequences. The analysis involved 53 nucleotide sequences. Bootstrap values (1000 replicates) are shown next to the branches. All positions containing gaps and missing data were eliminated. E. coli K12 (NC_000913.3) was used as an outgroup. Evolutionary analyses were conducted in MEGA7.
A phylogenetic tree based on core genome sequences was constructed using EDGAR 2.0. and is shown in Figure 2A, where Bt m401 was located in the same cluster with Bt sv. kumamotoensis. From the results obtained in the core genome tree, we selected the closest serovars (n=13) to compare the average nucleotide identity calculations (ANIb) (Fig. 2B). The analysis reinforces the high similarity between Bt m401 and Bt sv.kumamotoensis, with an ANIb value of 98.11%, followed by Bt sv. londrina (97.66%), Bt sv. indiana (97.51%), Bt sv. japonensis (97.44%), Bt sv. kurstaki (97.26%), Bt sv. coreanensis (97.24%), Bt sv. yunnanensis (97.2%), and Bt sv. galleriae (97.11%) (Fig. 2B). Based on both phylogenetic trees, we conclude that Bt m401 belongs to serovar kumamotoensis.
 Phylogenetic analyses based on bacterial core genomes of strains belonging to the Cereus clade. The tree for 42 genomes was built out of a core of 2034 genes per genome, 85428 genes in total. The numbers at the branching points are local support values computed by FastTree software using the Shimodaira-Hasegawa test (×100). Only values other than 100 are shown. (B) ANIb matrix for comparison of selected genomes retrieved from GenBank (accession numbers in parentheses). ANIb value calculations, clustering, and visualization as heat maps were done through EDGAR 2.0 web server.")
(A) Phylogenetic analyses based on bacterial core genomes of strains belonging to the Cereus clade. The tree for 42 genomes was built out of a core of 2034 genes per genome, 85428 genes in total. The numbers at the branching points are local support values computed by FastTree software using the Shimodaira-Hasegawa test (×100). Only values other than 100 are shown. (B) ANIb matrix for comparison of selected genomes retrieved from GenBank (accession numbers in parentheses). ANIb value calculations, clustering, and visualization as heat maps were done through EDGAR 2.0 web server.
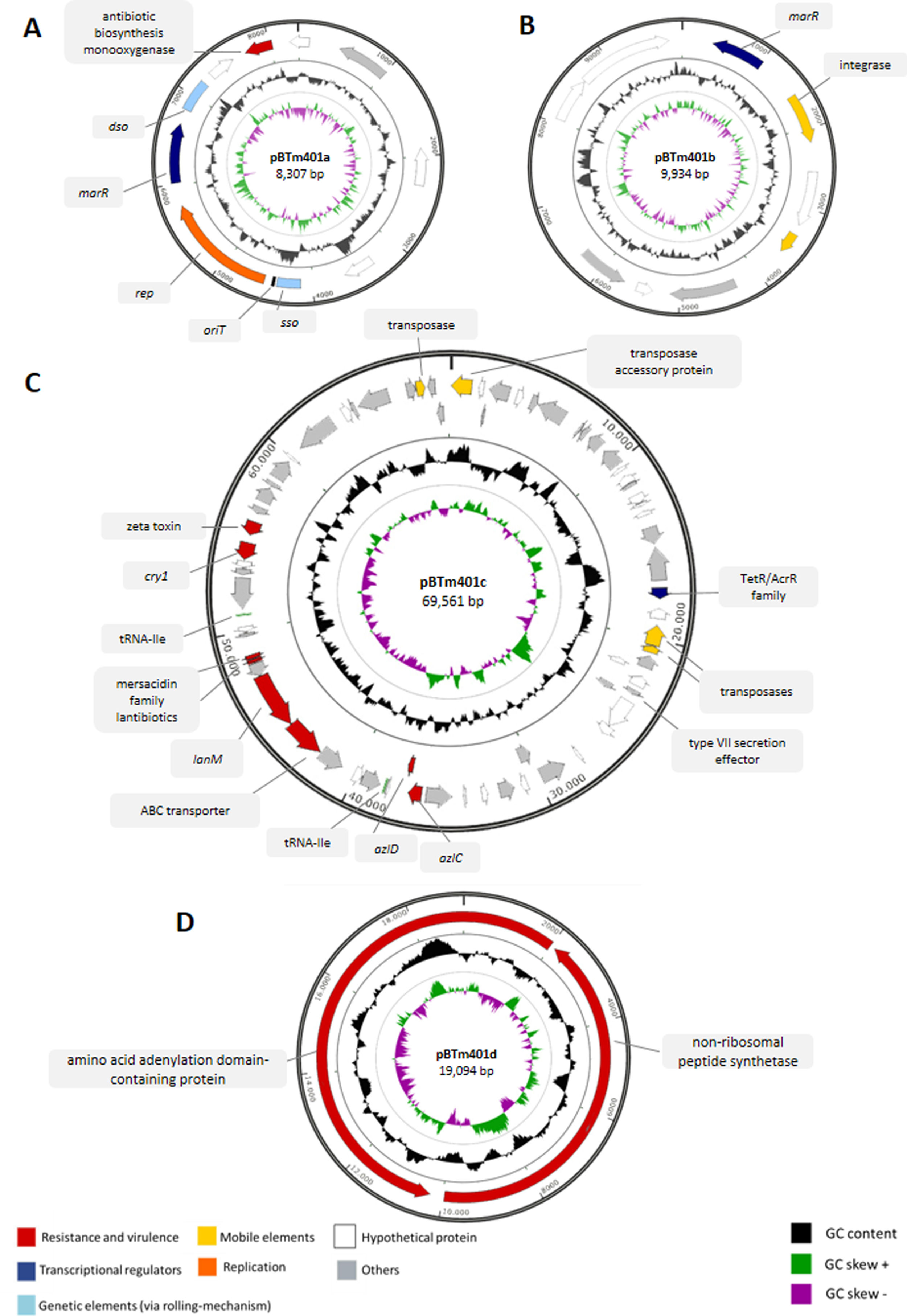
The complete sequences of plasmids pBTm401a, pBTm401b, pBTm401c, and pBTm401d were analyzed using different bioinformatics tools. A graphical representation is shown in Fig. S1, and their main characteristics are listed in Tables S2 and S3.
Plasmid pBTm401a (8307bp) presented genes coding a multiple antibiotic resistance regulator (MarR), an antibiotic biosynthesis monooxygenase, and a plasmid recombination protein (Table S3). The remaining genes were predicted as hypothetical proteins. Furthermore, pBTm401a contains genetic elements typical of plasmids replicating via the rolling-circle mechanism (RCR)10 (Fig. S1A). The single-strand origin (sso) is located upstream of the mobilization gene (4123–4378bp), and the double-strand origin (dso) is located downstream from the MarR and recombinase genes (6828–7179bp). Both dso and sso were identified by homology sequence with corresponding regions of other plasmids, such as pDx14.2 from B. mycoides (AJ871638) (82% identity for sso and 91% for dso) and pFR12 from Bt INTA-FR7-4 (EU362917) (83% identity for sso and 92% identity for sso). The origin of the transfer site (oriT) is located between the sso and the recombinase genes (4401–4430bp) and showed 90% homology with the oriT of plasmid pTX14-3 (X56204.1) from Bt sv. israelensis. The best hits obtained after BLASTn analysis of pBTm401a were selected for the individual comparison of the plasmids; we found partial similarities between pBTm401a and plasmid pFR12 from Bt strain INTA-FR7-4 (EU362917) (Fig. S2A), plasmid unnamed 32 from B. cereus NJ-W (CP012485), and plasmid pDx14.2 from B. mycoides (AJ871638), respectively. The conserved region between them corresponds to the predicted marR transcriptional regulator, the recombinase gene, and the genetic elements sso and dso.
The BLASTx analysis of plasmid pBTm401b (9934bp) showed only three putative protein products that codify a uracil permease, a MarR family transcriptional regulator, and an integrase (Fig. S1B and Table S3). The BLASTn analysis showed partial similarities with plasmids reported in Bt strains. The region comprising approximately from 2kb to 7kb in pBTm401b presented high homology with plasmid pBT1850012 from B. thuringiensis strain Bt185 (GenBank, CP014288) (Fig. S2B), plasmid p5 from B. thuringiensis strain QZL38 (GenBank, CP032611) and plasmid unnamed2 from B. cereus strain 09 (GenBank, CP042876). The sequence between 1.5kb and 6kb of pBTm401b presented high similarity with plasmid unnamed 8 from B. mycoides strain Gnyt1 (GenBank, CP020751).
Plasmid pBTm401c (69591bp) encodes two genes of mersacidin family lantibiotics, a lanthipeptide synthetase LanM, and an ABC transporter consecutively located (Fig. S1C and Table S3). Other relevant genes are summarized in Table S3, including a cry1 gene that showed 99% identity with cry1 described in megaplasmid poh1 of Bt ATCC 1079216. The best BLASTn hits for pBTm401c correspond to plasmids from strains of the Cereus clade: plasmid pBTm401c showed several small areas similar to those detected in plasmid p1 from B. thuringiensis strain HM-311 (CP040783) (97.3% identity), plasmid pBT1850636 from B. thuringiensis strain Bt185 (CP014283) (98.9% identity), plasmid pBb from Bacillus bombysepticus (CP007513) (98.6% identity), and plasmid unnamed1 from B. cereus strain 09 (GenBank, CP042875) (97.6% identity) (Fig. S2C). We found many similar regions in plasmid unnamed1 from B. cereus strain 09 (97.6% identity), including the 40–50kb region, which codifies lantibiotic genes.
Finally, pBTm401d (19094bp) analysis showed three putative protein products, a non-ribosomal peptide synthetase, and two amino acid adenylation domain-containing proteins (Fig. S1D and Table S3).
In addition, a set of genes associated with virulence factors were detected in Bt m401. In the chromosome, we found the operon hblABCD that encodes the hemolytic enterotoxin HBL. This feature correlates with a positive hemolytic phenotype in a blood agar test. These proteins are commonly found in strains of the Cereus clade, including Bt strains27. Other virulence factors found in the chromosome were the non-hemolytic enterotoxin complex, cytotoxin K, and enterotoxin FM (Table S4). The presence of sequences associated with virulence genes cytK, nheA, nheB, nheC, hblA, hblB, hblC, and hblD was confirmed by PCR (data not shown).
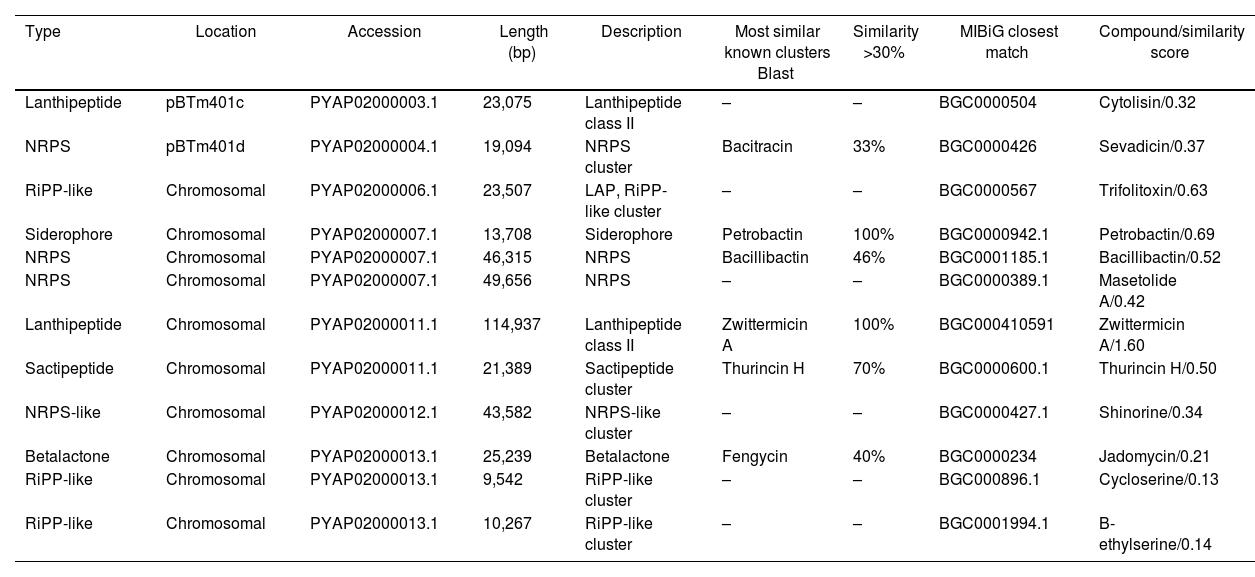
Prediction of biosynthetic gene clusters in the genomeBiosynthetic gene clusters (BCGs) in the genome and their corresponding secondary metabolites were predicted using the bacterial version of antiSMASH 6.0. The server predicted 12 regions of BGCs in the genome related to secondary metabolism, including bacteriocins, siderophores, ribosomally synthesized post-translationally modified peptide product clusters (RiPP-like), and non-ribosomal peptide synthetase clusters (NRPS) (Table 2).
Predicted biosynthetic gene clusters (BGCs) characterization in Bacillus thuringiensis m401 with antiSMASH 6.01 database and MIBiG repository
| Type | Location | Accession | Length (bp) | Description | Most similar known clusters Blast | Similarity >30% | MIBiG closest match | Compound/similarity score |
|---|---|---|---|---|---|---|---|---|
| Lanthipeptide | pBTm401c | PYAP02000003.1 | 23,075 | Lanthipeptide class II | – | – | BGC0000504 | Cytolisin/0.32 |
| NRPS | pBTm401d | PYAP02000004.1 | 19,094 | NRPS cluster | Bacitracin | 33% | BGC0000426 | Sevadicin/0.37 |
| RiPP-like | Chromosomal | PYAP02000006.1 | 23,507 | LAP, RiPP-like cluster | – | – | BGC0000567 | Trifolitoxin/0.63 |
| Siderophore | Chromosomal | PYAP02000007.1 | 13,708 | Siderophore | Petrobactin | 100% | BGC0000942.1 | Petrobactin/0.69 |
| NRPS | Chromosomal | PYAP02000007.1 | 46,315 | NRPS | Bacillibactin | 46% | BGC0001185.1 | Bacillibactin/0.52 |
| NRPS | Chromosomal | PYAP02000007.1 | 49,656 | NRPS | – | – | BGC0000389.1 | Masetolide A/0.42 |
| Lanthipeptide | Chromosomal | PYAP02000011.1 | 114,937 | Lanthipeptide class II | Zwittermicin A | 100% | BGC000410591 | Zwittermicin A/1.60 |
| Sactipeptide | Chromosomal | PYAP02000011.1 | 21,389 | Sactipeptide cluster | Thurincin H | 70% | BGC0000600.1 | Thurincin H/0.50 |
| NRPS-like | Chromosomal | PYAP02000012.1 | 43,582 | NRPS-like cluster | – | – | BGC0000427.1 | Shinorine/0.34 |
| Betalactone | Chromosomal | PYAP02000013.1 | 25,239 | Betalactone | Fengycin | 40% | BGC0000234 | Jadomycin/0.21 |
| RiPP-like | Chromosomal | PYAP02000013.1 | 9,542 | RiPP-like cluster | – | – | BGC000896.1 | Cycloserine/0.13 |
| RiPP-like | Chromosomal | PYAP02000013.1 | 10,267 | RiPP-like cluster | – | – | BGC0001994.1 | B-ethylserine/0.14 |
Abbreviations: LAP: linear azol(in)e containing peptide; RiPP-like: other unspecified ribosomally synthesized and post-translationally modified peptide product cluster; NRPS: non-ribosomal peptide synthetase cluster.
We investigated the antagonistic potential of Bt m401 against 52 strains of P. larvae using a well diffusion technique. Clear inhibition zones were observed when testing Bt m401 against all the strains tested, ranging from 17.33±0.47 to 32.67±0.47mm for genotypes ERIC I; from 14.67±0.47 to 24.33±0.47mm for genotypes ERIC II, and from 14±0.82 to 21.67±0.47mm for genotypes ERIC IV, respectively at 24h (Table 1). Interestingly, all the tetracycline-resistant strains of P. larvae (i.e., PL373, PL374, PL391, PL394, PL395, PL442, and PL443) were inhibited by Bt m401.
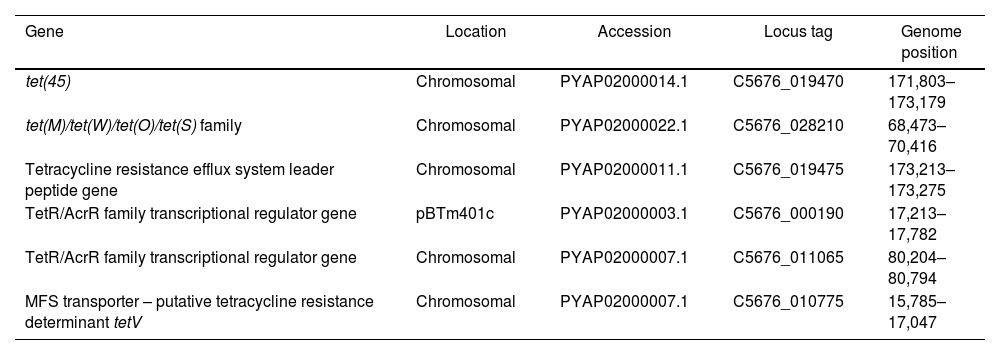
Tetracycline resistance genesTetracycline-resistance genes and relatives detected in Bt m401 are summarized in Table 3. Bt m401 owns two TETR genes corresponding to tet(45) and tet(M)/tet(O)/tet(S) family, both located in the chromosome. The best hits of the BLASTn analysis revealed high identity homology (>95%) with megaplasmids from Bt strains, i.e., plasmid p1 from HM-311 (CP040783.1), plasmid pBT62A from BT62 (CP044979.1), plasmid pBT1850636 from Bt185 (CP014283.1), and the megaplasmid poh1 from Bt ATCC 1079216. The presence of these genes in the Bt m401 chromosome led us to suspect that this genome region could be part of an integrated plasmid. Additionally, we found a putative tetracycline resistance determinant tet(V) located in the chromosome with high identity homology (99%) with B. cereus VD140 (GCA_000399545).
Tetracycline-resistance genes in Bacillus thuringiensis m401
| Gene | Location | Accession | Locus tag | Genome position |
|---|---|---|---|---|
| tet(45) | Chromosomal | PYAP02000014.1 | C5676_019470 | 171,803–173,179 |
| tet(M)/tet(W)/tet(O)/tet(S) family | Chromosomal | PYAP02000022.1 | C5676_028210 | 68,473–70,416 |
| Tetracycline resistance efflux system leader peptide gene | Chromosomal | PYAP02000011.1 | C5676_019475 | 173,213–173,275 |
| TetR/AcrR family transcriptional regulator gene | pBTm401c | PYAP02000003.1 | C5676_000190 | 17,213–17,782 |
| TetR/AcrR family transcriptional regulator gene | Chromosomal | PYAP02000007.1 | C5676_011065 | 80,204–80,794 |
| MFS transporter – putative tetracycline resistance determinant tetV | Chromosomal | PYAP02000007.1 | C5676_010775 | 15,785–17,047 |
MIC values for TET, OTC, and minocycline (MIN) were determined for Bt m401. The results obtained were MIC TET=128 (R), MIC OTC>128 (R), and MIC MIN=2 (S), respectively. Interestingly, in Bt m401, cross-resistance was found between TET and OTC but not with MIN. Values of MIC obtained for reference strains S. aureus ATCC 29213 and E. coli ATCC 25922 were within the tolerance range acceptable for quality control, according to CLSI standards17,18.
Visualization of plasmid DNAThe plasmid profile of Bt m401 showed two plasmids between 5000bp and about 8000bp and two larger plasmids of more than 19000bp (Fig. S3). These observations correlated with genome assembly data showing four plasmids of 8307bp, 9934bp, 19094bp, and 69591bp, respectively (Table S2).
PCR analysis of tetracycline-resistant genes and phylogenetic analysis of tet(45) and tet(L) genesResults of the PCR assays revealed that Bt m401 yielded the expected tet(45) product of about 107bp and also the tet(L) product of approximately 788bp, respectively, when using genomic DNA as a template (Fig. S4). However, no amplification was obtained when using plasmid DNA as a template. In contrast, no PCR products were detected using the tet(K), tet(M), tet(O), tet(W), otr(A), or otr(B) primers, neither with genomic nor plasmid DNA as a template. The phylogenetic analysis of the tet(45) gene of Bt m401 is shown in Fig. S5, confirming that it was more closely related to the tet(45) gene from Bacillus sp. 6f (KX091845.1).
DiscussionWe characterized the strain of B. thuringiensis m401 isolated from honey using several approaches. We performed an in-silico analysis of the Bt m401 genome, mainly focused on detecting genes encoding antibiotic resistance, antimicrobial peptides, toxins, and related regulatory gene factors. We also tested its capacity as a biocontrol agent against P. larvae, the causal agent of AFB disease of honeybees.
The bacterial colonies and media appearance in Bacillus Chromoselect agar and PEMBA correspond to those described for B. thuringiensis3,31. In addition, the results of morphological and biochemical tests and PCR/RFLP analysis of genes encoding 16S rRNA match those reported for Bacillus thuringiensis3,32. The identity of the strain was confirmed as Bacillus thuringiensis sv. kumamotoensis based on the average nucleotide identity calculations (ANIb) comparison and the phylogenetic analysis of the gyrB nucleotide sequences.
The genome of Bt m401 contained four plasmids of 8307bp, 9934bp, 19094bp, and 69591bp, respectively. Other authors also reported various plasmids and megaplasmids in Bt strains9,16,23,27. The analysis of plasmid sequences showed that pBTm401a and pBTm401b contain coding one-component signal transduction regulators such as MarR, while pBTm401c contains a transduction regulator TetR/AcrR. Plasmids containing signal transduction regulators have been reported in Bt, e.g., plasmids pFR12.5 and pFR55 from Bt INTA-FR7-4 that codify a putative transcriptional regulator MerR9 and Bt ATCC 10792 that possess a megaplasmid poh1 containing a TetR/AcrR coding gene16. The target genes of these transcriptional regulators involve various critical functions, including osmotic stress, efflux pumps, multi-drug resistance, and virulence19.
The organization of replication elements in pBTm401a is similar to corresponding regions of pTX14-3 and other plasmids belonging to RCR group VII10. The mob gene (mob14-3) sequence from plasmid pTX14-3 showed 86% homology with the recombinase gene of pBTm401a, and the replication p14-3 gene showed 76% homology with marR transcriptional regulator of pBTm401a.
Plasmid pBTm401c encodes two genes of mersacidin family lantibiotics and a lanthipeptide synthetase (Tables 2 and S3). Lanthipeptides are a class of ribosomally synthesized and post-translationally modified peptides (RiPP) containing characteristic thioether cross-links imperative for bioactivity and stability, and those that possess antibacterial activity are called lantibiotics28. Lantibiotics have been extensively studied for their broad-range activity against clinically relevant pathogens. Several Bacillus species, including Bt, produce lantibiotics, e.g., clausin in B. clausii, formicin in B. paralicheniformis APC 1576, ericins in various strains of B. subtilis, haloduracin in B. halodurans strain C-125; thusin in B. thuringiensis strain BGSC 4BT1; lichenicidins in B. licheniformis VK21 and I89 strains and mersacidin in B. amyloliquefaciens FZB42 and B. subtilis HIL Y-85,547282,28,29. Another lipopeptide biosynthetic gene cluster followed by a transposase gene was detected in the 312kb plasmid from B. thuringiensis BMB17149. The presence of lantibiotic genes encoded on plasmids and flanked by transposases allows to infer a possible mobilization of these elements among bacterial strains. Other relevant genes found in pBTm401c codify for a zeta toxin, two azaleucine resistance proteins AzlC/AzlD, a type VII secretion system, and a cry1 gene (Tables S3 and S4). The presence of a putative cry gene that codifies parasporal crystals correlates with the crystals observed by optical microscopy. The parasporal crystal protein Cry is the primary insecticidal toxin of B. thuringiensis35. It has been reported that some Bt strains produce Vip and Sip insecticidal proteins37; however, we did not find any homologous sequence to sip or vip genes that codify, respectively, to Vip and Sip protein families.
On the other hand, plasmid pBTm401d contains a non-ribosomal peptide synthetase cluster (NRPS) (Tables 2 and S3). The presence of non-ribosomally peptide synthetases is a relevant characteristic; several Bacillus species produce antimicrobial peptides mediated by these synthetases; typical examples are surfactins, iturins, fengycines, and pliplastatins43. Surprisingly, pBTm401d showed high similarities with chromosomal regions of Bt strains HM-311 (CP040782) (99.2% identity), QZL38 (CP032608), and Bt185 (CP014282) (99.2% identity). This particular sequence exhibits repetitive regions that may interfere with assembling programs, suggesting an incorrect or incomplete genome assembly in those Bt strains. Besides, the whole sequence of pBTm401d showed similarity with a partial region of plasmid p1 from B. cereus strain CTMA 1571 (CP053657) (Fig. S2D), where this region is located between IS110 and IS3 family transposases. The presence of transposases suggests a possible horizontal transmission of the target region among different bacterial strains. The similarities among plasmid sequences of several strains of the Cereus clade, including B. thuringiensis, imply an active horizontal transfer of extrachromosomal elements in nature.
The genome mining analysis revealed 12 regions of biosynthetic gene clusters responsible for synthesizing secondary metabolites. We identified biosynthetic gene clusters coding for bacteriocins, siderophores, ribosomally synthesized post-translationally modified peptide products, and non-ribosomal peptide synthetase clusters. We found a BGC coding for a lantipeptide class II in plasmid pBTm401c and a BGC coding for an NRPS with 33% similarity to bacitracin in plasmid pBTm401d. Bacitracin is a cyclic cationic, non-ribosomally synthesized dodecapeptide reported in different Bacillus species, with activity against Gram positive bacteria and fungi43. Within the Cereus clade, bacitracin genes have been identified in Bt and B.weihenstephanensis27. As previously described, plasmid pBTm401c encodes two consecutive genes of mersacidin family lantibiotics, a lantipeptide synthetase LanM, and an ABC transporter. Mersacidin belongs to Type-B lantibiotics, and targets cell wall precursor lipid II and inhibits cell wall synthesis.
In the chromosome, we detected BCG coding for petrobactin (100%), zwittermicin A (100%), thurincin H (70%), bacillibactin (46%), and fengycin (40%). Zwittermicin A is a linear cationic lipopeptide with potent antibiotic and antifungal activity reported in B.thuringiensis20,27 and B. cereus42. Thurincin H is a bacteriocin belonging to class II (non-modified peptides), subclass II.2 (thuricin-like peptides) according to Abriouel and co-workers2, and recently reclassified as a sactipeptide8. Thurincin H has been reported in Bt serovars with antimicrobial activity against a wide range of Gram positive bacteria15,30. Fengycin is a cyclic non-cationic lipopeptide non-ribosomally synthesized reported in Bt strain SM1 with antifungal activity against Candida albicans40. Other Bacillus species also produce fengycins with potent inhibition against pathogenic filamentous fungi36. The presence of catecholate siderophores, such as petrobactin and bacillibactin, in Bt m401 suggests iron acquisition abilities. This siderophore can potentially promote plant growth11. Other authors reported gene clusters assigned to the production of petrobactin and bacillibactin in different Bt strains20,27. These gene clusters are not exclusive to Bt since they are also found in other species belonging to the Cereus clade11,20.
Clear inhibition zones ranging from 14±0.82 to 33.00±1.41mm at 24h (Table 1) were observed when testing Btm401 against all P. larvae strains evaluated. Previous studies indicated that inhibition of P. larvae was strain-dependent6,12,34. In contrast, Bt m401 showed inhibition against all P. larvae strains tested in vitro (n=52) belonging to different genotypes and geographical origins.
Antibiotic resistance determinants are also a common part of the genomes of environmental bacteria. In most bacteria, tetracycline resistance is due to acquiring new genes, often associated with mobile elements39. The PCR assays revealed that Bt m401 yielded both tet(45) and tet(L) products when using genomic DNA as a template (Fig. S4). Due to the high similarity between the tet(45) and tet(L) sequences selected to design primers for specific PCRs47,48, we obtained the expected amplicons in both cases. Nevertheless, BLASTn analysis revealed that the nucleotide sequence presented here and located in the chromosome (PYAP02000014.1) showed higher similarities to tet(45) gene from Bacillus sp. 6f (96% identity) (KX091845.1), which was later identified as B. cereus strain 6f50; tet(45) gene from Escherichia coli strain DH5alpha (94%) (GU584222.2), and tet(45) gene from Bhargavaea cecembensis strain DMV42A (93% identity) (GU584217.2) than to the sequence of tet(L) from plasmid pBHS24 from Bacillus sp. 24 (80% identity) (HM235948.1); tet(L) from plasmid pERGB of Staphylococcus aureus strain 69371 (80% identity) (JN970906.1); and tet(L) gene f Bacillus sp. DMV3A (78% identity) (JN232537.1) (Fig. S5). As far as we know, within Bacillus species, the tet(45) determinant was only detected in seven strains of B. cereus and one strain of Bt, respectively50. Based on these results, we concluded that Bt m401 harbored the tet(45) resistant gene in the chromosome.
ConclusionsIn this work, we identified a tetracycline-resistant strain of Bacillus thuringiensis sv. kumamotoensis (Bt m401) isolated from honey based on the average nucleotide identity calculations (ANIb) comparison and the analysis of the gyrB gene sequences of different B.thuringiensis serovars. The insights on the genome analysis revealed sequences with homology to virulence factors [cytK, nheA, nheB, nheC, hblA, hblB, hblC, hblD, entFM, and inhA]; tetracycline resistance genes [tet(45), tet(V), and tet(M)/tet(W)/tet(O)/tet(S) family], and regions of biosynthetic gene clusters coding for bacteriocins, siderophores, ribosomally synthesized post-translationally modified peptide products, and non-ribosomal peptide synthetase clusters that provide evidence for the possible use of Bt m401 as a biocontrol agent. Moreover, Bt m401 displays antagonistic activities against different genotypes of P. larvae, the causal agent of AFB of honeybees. The production of antagonistic compounds and resistance to tetracycline and oxytetracycline contributes to extending its host range and virulence. These findings provide a valuable background for developing biotechnological and biocontrol applications for Bt m401, which should be addressed in future experiments.
Data availabilityThis Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession PYAP00000000. The version described in this paper is version PYAP00000000. 2. Plasmids sequences have been deposited at DDBJ/EMBL/GenBank under accession numbers PYAP02000001.1 for pBTm401a; PYAP02000002.1 for pBTm401b, PYAP02000003.1 for pBTm401c, and PYAP02000004.1 for pBTm401d, respectively. The data in this article are available in the paper and its online supplementary material.
Authors’ contributionsAMA designed the experimental work, contributed to microbiological procedures and data analysis, wrote and reviewed the manuscript, and provided funding. FL completed molecular biology experiments and data analysis. EA and ACL performed experimental work. GATT executed data analysis and graphic constructions using EDGAR. All authors have read and approved the final version of the manuscript.
Conflict of interestThe authors declare that they have no conflicts of interest.
AMA is a Member of the Scientific Research Career of CICBA, Argentina; ACL and GATT are members of the Scientific Research Career of CONICET, Argentina, and EA is a post-doctoral fellow of CONICET, respectively. The authors acknowledge the collaboration of Dr. Jochen Blom for helping with the EDGAR platform. This work was supported by the ANPCyT, Argentina (Grant PICT 2017-2014 to AMA) and Comisión de Investigaciones Científicas (CIC) de la Provincia de Buenos Aires, Argentina (Grant No. 389/21, res. 1538/21).
The following are the supplementary data to this article:

Comparative alignment of Bt m401 plasmids with related plasmids. The best BLASTn hits for each plasmid are displayed on Kablammo's online server. Comparisons between: (A) pBTm401a and pFR12 from Bt INTA-FR7-4. (B) pBTm401b and pBT1850012 from Bt185. (C) pBTm401c and unnamed plasmid 1 from B. cereus 09. (D) pBTm401d and p1 from B. cereus CTMA 1571. Preserved regions are paired with shaded regions. The intensity of color indicated the strength of the match. GenBank accession numbers in parentheses.
Plasmid pattern of Bt m401 in 0.5% agarose gel. Lanes: (1) Paenibacillus larvae PL373 used as a marker for plasmid sizes and (2 and 3) B. thuringiensis m401.
Agarose gel electrophoresis of DNA fragments generated by PCR amplification of total genomic DNA of B. thuringiensis m401 strain with tet(L)-F/tetL-R (lane 1), and tet(45)F/tet(45)/R (lane 2) primers. Lane M, DNA molecular weight marker 100–1000 bp (Inbio Highway®, Argentina), lane 3, nontemplate control.
Phylogenetic tree of tet(45) and tet(L) genes. Nucleotide sequences were aligned using Clustal W, and the tree was constructed with MEGA 7 using the ML method based on the General Time Reversible model. Bootstrap values (1000 replicates) are indicated at the nodes. The analysis involved 14 nucleotide sequences. All positions containing gaps and missing data were eliminated. The amino acid sequences of the tet(M) gene from Clostridium difficile strain CD2386 (JN846696.1) were used as an outgroup.
Global statistics of Bt m491.
General overview of plasmids pBTm401a, pBTm401b, pBTm401c, and pBTm401d.
Most relevant genes and genetic elements found in the plasmids of B. thuringiensis m401.
Predicted virulence factors of B. thuringiensis m401.
