



Polyarteritis nodosa (PAN) is a necrotizing vasculitis, rare in childhood, and characterized by the inflammation of small and medium vessels and multiple organ involvement. The case is presented of a 4-year old girl with prolonged febrile syndrome, chronic abdominal pain, disabling myalgia, and skin involvement. After 2 years of symptoms, she met clinical criteria for PAN. She received treatment with oral and intravenous systemic corticosteroids, 6 months of intravenous cyclophosphamide and maintenance with conventional immunosuppressants without an adequate response. However, she showed clinical improvement with oral cyclophosphamide and long-term corticosteroids. She had several relapses during follow-up visits due to irregular treatment requiring a high cumulative dose of cyclophosphamide. Five years later she presented with macroscopic haematuria, and was assessed for, among other causes, cyclophosphamide toxicity and disease activity. The workup included cystoscopy and bladder biopsy with finding of necrotizing vasculitis of bladder wall. Bladder vasculitis is rarely reported in the literature (3–5 cases in adults) and in that consulted there are no reports in children. To our knowledge, this is the first case of bladder involvement associated with systemic vasculitis reported in the pediatric age.
La poliarteritis nudosa (PAN) es una vasculitis necrosante, rara en la infancia, caracterizada por el compromiso de vasos pequeños/medianos y de múltiples órganos. Presentamos a una paciente que inició a los 4 años con síndrome febril prolongado, dolor abdominal crónico, mialgias incapacitantes y compromiso en la piel, quien luego de 2 años de cuadro clínico completa criterios clínicos para PAN. Recibió tratamiento con corticoide sistémico por vía oral e intravenosa, 6 meses de ciclofosfamida por vía intravenosa y manejo de mantenimiento con inmunosupresores convencionales sin respuesta adecuada, logrando control de la enfermedad únicamente con ciclofosfamida por vía oral y corticoide a largo plazo. Luego de 5 años y de recibir una dosis alta acumulada de ciclofosfamida, inicia con cuadros de hematuria macroscópica. Se evaluaron, entre otras causas, la toxicidad por ciclofosfamida y la actividad de la enfermedad. El estudio incluyó biopsia vesical, con hallazgo de vasculitis necrosante de paredes vesicales. La vasculitis vesical es raramente reportada en la literatura (3–5 casos en adultos) y en lo consultado no hay reportes en niños. Se describe, en nuestro conocimiento, el primer caso de compromiso vesical asociado a vasculitis sistémica reportado en la edad pediátrica.
Polyarteritis nodosa (PAN) was described for the first time by Kussmaul and Maier in 1866.1 It is defined as a necrotizing vasculitis of medium/small vessels, with no vasculitis in the arterioles, venules or capillaries, and is not associated with antineutrophilic cytoplasmic antibodies (ANCA).2 This is the most frequent vasculitis in children, following IgA vasculitis/Henoch-Schönlein purpura and Kawasaki's disease.3 In adults, the incidence of PAN is around 0.0–1.6 cases, while its prevalence is approximately 31 cases per million4; it is less frequent in infants, although there are very few epidemiological data in children.5 In the pediatric population there is a peak of the disease at around 9–10 years, and both genders are equally affected.6,7 Adult PAN is often associated with hepatotropic viral infections, while in children streptococcus and a few viruses have been suggested as potential causes, with no conclusive evidence.8,9
There are 2 forms of presentation in PAN during childhood. Cutaneous PAN, in most cases self-limiting, which can present with livedo reticularis, purpura, splinter hemorrhages, finger ischemia, painful nodules, arthritis, and myositis. Overall, the internal organs are not affected.8 Systemic PAN has a more severe spectrum of presentation and, in addition to the manifestations of cutaneous PAN, patients may experience abdominal pain and intestinal angina, which in some cases progress to intestinal bleeding.3,8 The involvement of the renal vessels leads to hypertension and renal failure, and rarely testicular pain and infarction are also present; at the neurological level, the peripheral nervous system is often compromised in the form of multiple mononeuritis, whilst the central nervous system is usually not affected. Ocular involvement has also been described, while in general there is no pulmonary involvement, which helps to differentiate PAN from other types of vasculitis.
PAN mortality is low (around 4%) and its evolution may be total remission or relapse–remission.3,6–8
Laboratory tests are non-specific; during the activity of the disease, there may be leukocytosis, thrombocytosis, and acute phase reactants elevation. Autoantibodies are usually negative, including ANCA.10
Pediatric PAN is classified in accordance with the EULAR/PRINTO/PRES criteria revised in 201011; it should be highlighted however, that these are classification and not diagnostic criteria. According to these criteria, the typical histopathology of necrotizing vasculitis or angiographic anomalies are essential for making a diagnosis, with a sensitivity of 84 and 81%, respectively, and a specificity of 99% for both.11 The clinical findings include skin involvement (livedo reticularis, subcutaneous nodules, superficial and profound infarctions, minor ischemic changes, necrosis/gangrene), muscular pain or sensitivity, hypertension (systolic/diastolic blood pressure >95 percentile), sensory peripheral neuropathy or mononeuritis and renal involvement presenting with proteinuria, hematuria and glomerular filtration rate alteration.11
Although the conventional angiography continues to be the “gold standard” and has the highest sensitivity to detect stenosis of the smallest vasculature and microaneurysms, it is an invasive technique with a significant exposure to high radiation doses. This is the reason why MRI angiography and CT angiography are increasingly being used in children.11,12
Most treatments for children therapies are extrapolated from adult essays.13 Induction therapy with glucocorticoids and cyclophosphamide (CYC) is used, and maintenance therapy with oral azathioprine (AZA), methotrexate or leflunomide. The use of biologics in retrospective cohorts has proven to be efficient and safe in children with primary systemic vasculitis. However, treatment protocols and clinical trials are still required.14,15
Clinical caseThis is a 4-year old female patient, previously healthy and uneventful medical history, with an initial presentation of fever and odynophagia, in addition to a diagnosis of bacterial tonsillitis (Streptococcus was not confirmed). The patient received conventional outpatient antibiotic therapy.
During the following 6 months, the patient visited the emergency department on several occasions because of 3–4 days fever of 38–39°C, associated mostly with postprandial abdominal pain and sometimes disabling pain of the lower extremities. The first line tests conducted were normal, though occasionally the blood test showed mild leukocytosis and thrombocytosis, acute phase reactants elevation, mainly ESR, without overtly compromising the clinical condition, and so the patient received outpatient care.
After 8 months of the initial presentation, the patient developed prolonged fever over 22 days, accompanied by postprandial abdominal pain and intensification of myalgia which intermittently prevented ambulation and hence the patient had to be hospitalized. The physical examination identified recent onset Raynaud's phenomenon and livedo reticularis as the only findings.
A complete extension study was conducted, ruling out frequent and infrequent causes of infection; the bone marrow testing for malignancy was negative for blast, tumoral and hemophagocyte cells; the skin biopsy was compatible with livedo reticularis and the immunology test with negative antibodies and normal complement. After 26 days of normal tests, in light of the resolution of fever and adequate clinical status of the patient, the ambulatory tests were continued and these were also negative.
After one year of erratic follow-up, with 1–2 fever episodes per month, the patient began to experience reduced visual acuity in the right eye, and the ophthalmology exam identified optic nerve atrophy. The autoimmune trials were once again negative.
Two months later, the patient returned to the ER with fever, abdominal pain and headache with hypertensive emergency that required ICU admission for several days due to difficult to control high blood pressure (HBP) (the study for secondary causes of HBP was normal). The laboratory tests showed leukocytosis, thrombocytosis and significant elevation in acute phase reactants (ESR and CRP). During ICU admission the present developed mesenteric angina (it was impossible to do an angiography due to the patient's clinical condition). The analysis of the clinical manifestations and the severe systemic inflammatory response led to consider PAN-like systemic vasculitis as a potential diagnosis. Treatment was initiated with glucocorticoid pulses (3) and it was continued at 1mg/k/day associated with HBP management with 2 agents, on account of the difficulty to control blood pressure levels.
During follow-up, the patient received treatment with steroids at a dose of 1mg/k/day, presenting mild persistent activity with recurrences; on one occasion presented with renal involvement, so the decision was made to start IV CYC; the patient received 6 monthly doses and prednisolone at high doses (2mg/k/day), and remission was achieved. There were some difficulties with maintenance therapy because of poor compliance, methotrexate intolerance and AZA failure. The case was submitted to a pediatric rheumatology board and the decision was made to start oral CYC (2mg/kg/day), which led to the control of the disease.
After 5 years of onset of the disease (9 years old), follow-up is irregular because of the patients psychosocial environment (economic conditions, administrative formalities, poor compliance, etc.). The patient receives oral CYC on and off, and variable doses of prednisolone, with no effective response. At this point, the patient is experiencing hematuria and lower urinary tract symptoms. The study for hematuria indicates a non-glomerular etiology and low involvement; the renal ultrasound is normal. Due to the history of extended oral CYC use (despite doubts about compliance), an accumulated dose of 100g in 5 years, and the risk of CYC-associated side effects (hemorrhagic cystitis [HC]), the decision was made to discontinue CYC, reintroduce AZA and assess the response.
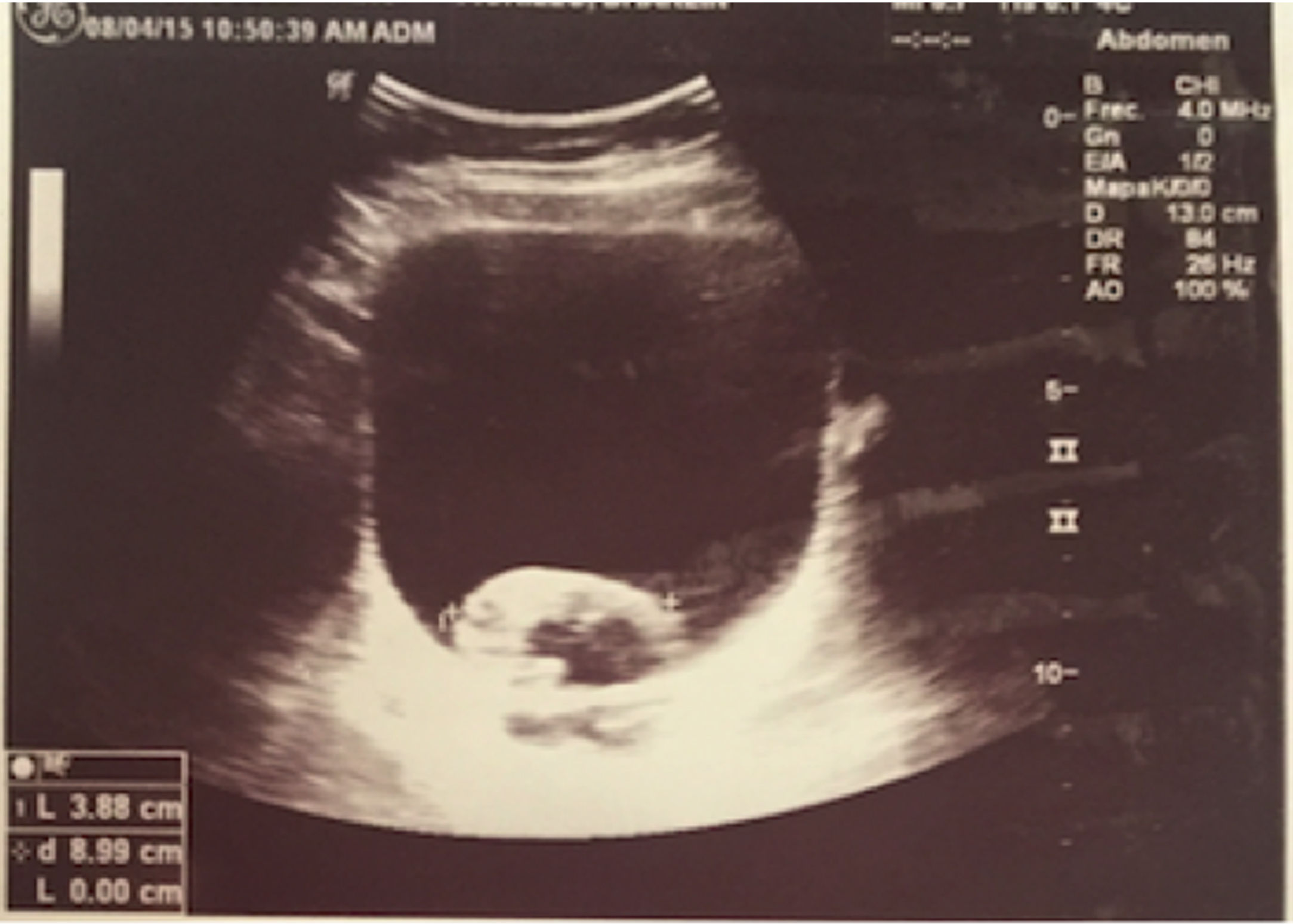
Notwithstanding the discontinuation of CYC, after 8 months, there are still relapsing episodes of hematuria, one of them was severe, compromising the hemodynamic status of the patient, with a drop in hemoglobin down to 7g/dl, in addition to the presence of a large intravesical clot (Fig. 1). Further pediatric nephrology and urology studies were conducted to identify other causes for the persistent hematuria, including poliomavirus, adenovirus, mycobacteria, interstitial nephritis, eosinophilic cystitis, etc., which were all negative. The uro-CT documented mild and diffuse thickening of the bladder walls, with negative results for infection and persistence of symptoms. The decision was made to conduct a cystoscopy which showed a hyperemic mucosa with generalized vasculitis in the vesical mucosa.

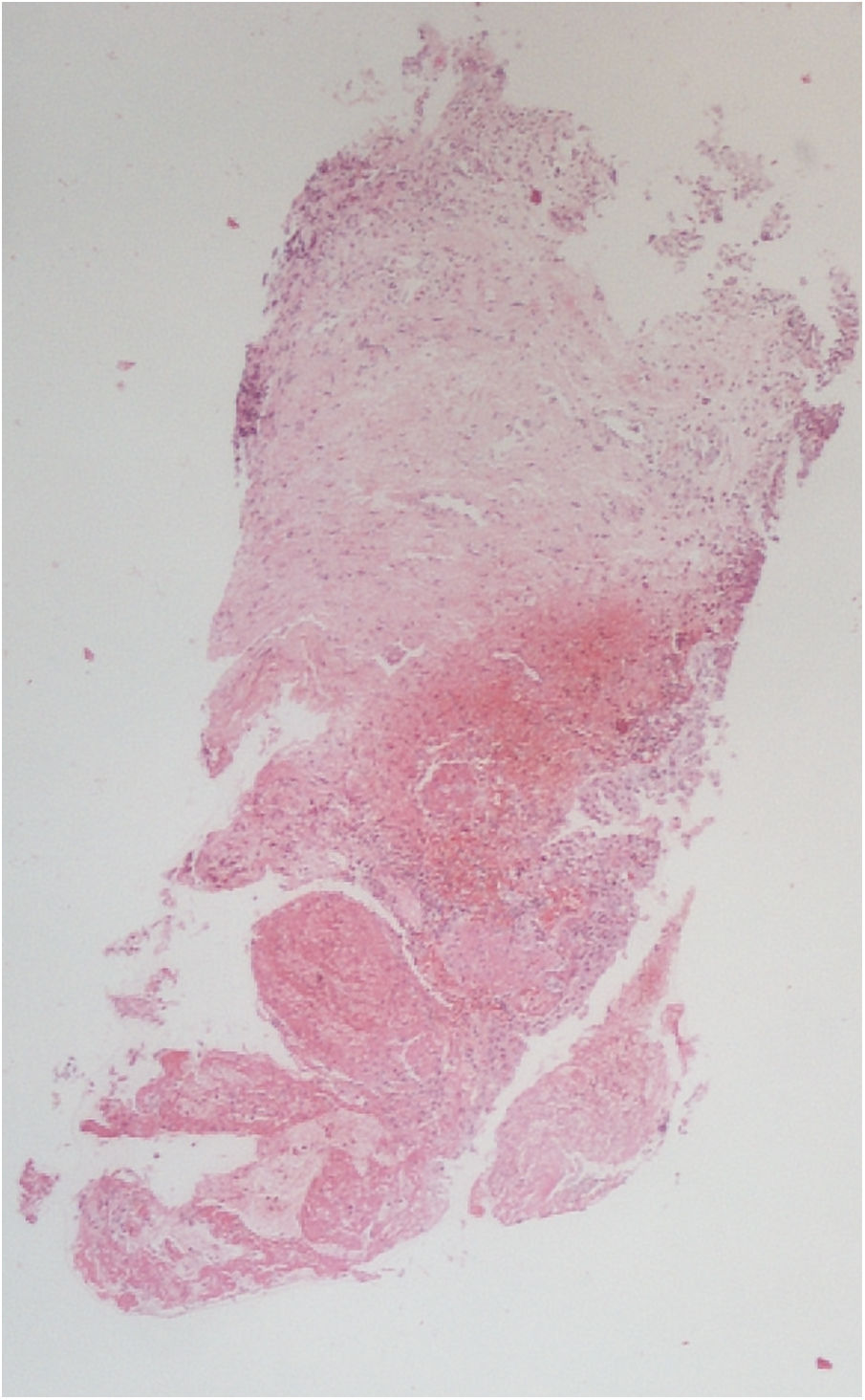
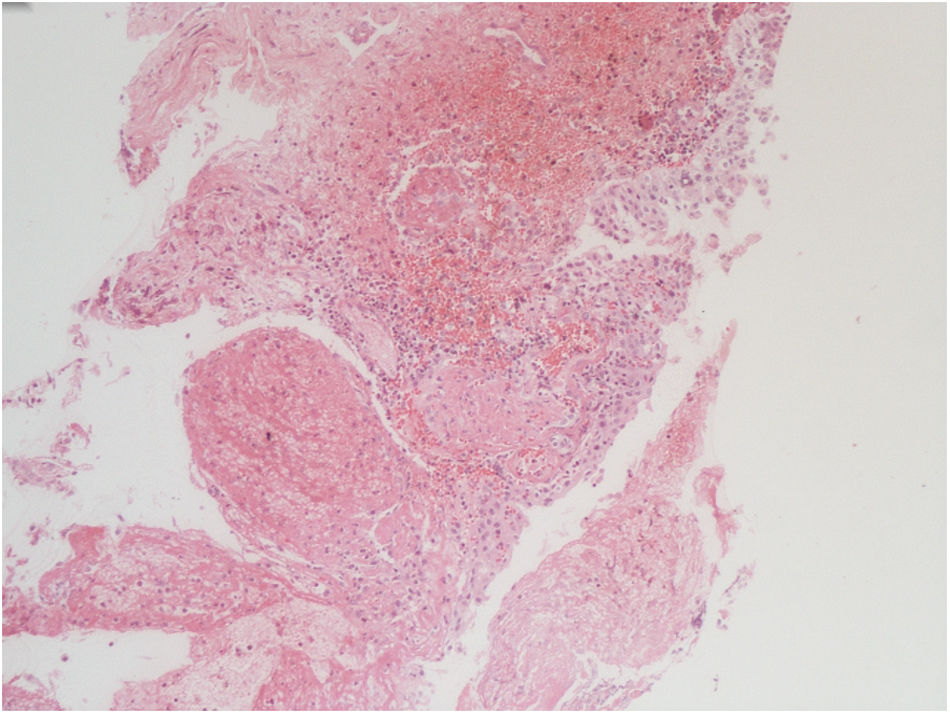
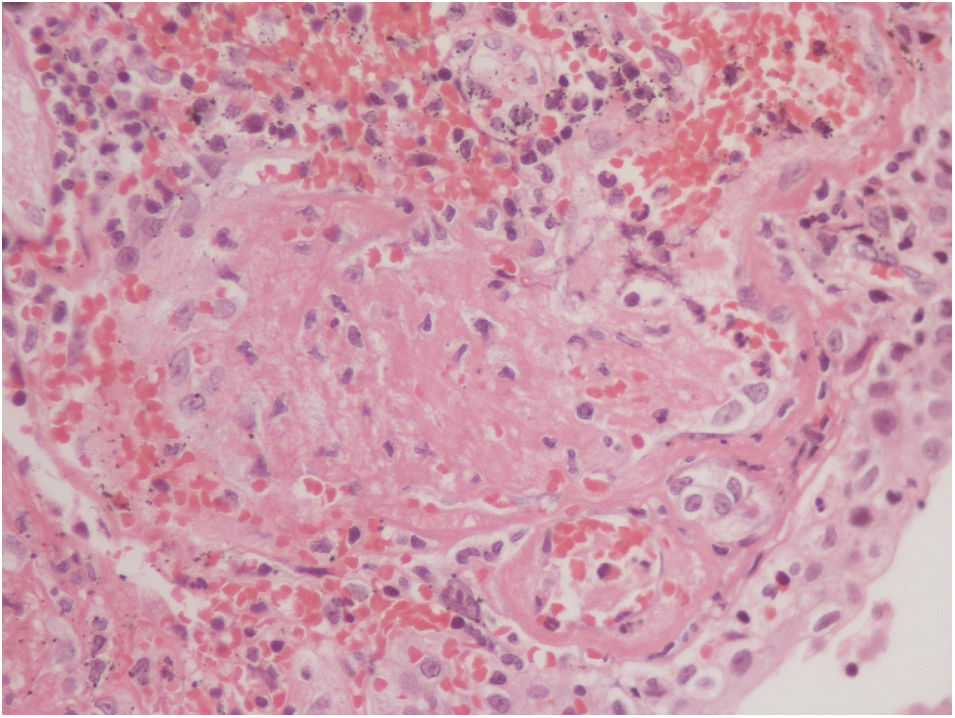
The vesical histopathology showed a predominant pattern of vasculitis damage (Figs. 2 and 3), with necrosis of the vascular walls, with associated endothelial edema and marked secondary leukocytoclastic inflammatory response, bleeding and necrosis (Fig. 4).



The unusual and severe compromise of this patient who was refractory to other treatments and received steroids for an extended period of time, led to reconsider therapeutic management. Once again, at a Rheumatology Board meeting and with the support of the literature (though sparse), treatment was initiated with rituximab at a dose of 750mg/m2 in weeks 0 and 2, associated with IV CYC 500mg and glucocorticoid, which managed to control de complication and achieved remission.
Currently the patient is 16 years old, her evolution has been favorable and she has tolerated AZA maintenance therapy, with no new episodes of hematuria, improved compliance, understanding and control of the disease. This made possible the progressive tapering of the steroid dose until its total discontinuation at present.
DiscussionPAN is considered an exclusion diagnosis, characterized by its multisystemic involvement, with symptoms that are frequent in other conditions (infection, malignancy, etc.),16 requires a high index of suspicion and represents a genuine diagnostic challenge for clinicians.
The characteristics of the condition vary depending on the organs involved,7 though any system may be involved. The clinical characteristics of a systemic PAN pediatric cohort in 609 children were as follows: fever (87%), myalgia/muscle sensitivity (83%), skin (88%), renal (19%), severe gastrointestinal (10%) and neurological compromise (10%).6 Hypertension is described in around 43–90% in the various cohorts of pediatric patients with PAN, and in general it is difficult to manage.6,7,17,18
A cohort with 22 children19 was reported in Colombia in 2017, in which the most frequent initial manifestations were joint involvement in 72% of patients, followed by constitutional symptoms including fever, myalgias (68%) and skin manifestations; the organic involvement was mostly renal and gastrointestinal, in the latter with severe manifestations such as intestinal angina.
Skin nodules are the most frequent skin manifestation in patients with juvenile PAN; they present as erythematous nodules in the lower extremities, measuring from 1 to 5cm in diameter and may coalesce to develop large areas of roughened skin.17 The appearance of livedo refers to a clinical characteristic where the reticular erythema extends over a large area17; it is defined as a reticular or mottled skin pattern, purple, red or lingering blue color, that does not revert with heat, in the trunk, arms, or legs. It is a regular smooth lesion, usually as continuous circles (livedo reticularis regular) or broken irregular circles (livedo racemosa), which in the latter case is associated with diseases such as antiphospholipid syndrome, vasculitis and scleroderma, inter alia.20 The cutaneous ulcerations in the lower extremities have a longer course and are related to neuropathy.17 The neurological condition has been reported in the literature as peripheral neuropathy, focal defects, hemiplegia, visual loss, multiple mononeuritis, renal nerve paralysis, organic psychosis and seizures.18,21 The severe GI involvement has been associated with a higher risk of relapse and can manifest itself as mesenteric infarction or angina, perforation, peritonitis, inter alia.22 The ocular manifestations may occur as the initial sign in about 10–20% of patients; inside the eye, there may be occlusion of the central retinal branch and artery, ischemic retinopathy, transient monocular visual loss, proptosis, bitemporal visual field defect, anterior or posterior ischemic optic neuropathies, and it is important to acknowledge that the vasculitis process is not limited to the retinal circulation: it may also show up as conjunctivitis, scleritis, uveitis or neuritis23; when the vascular supply to the optic nerve is affected (primarily the choroidal vessels and the posterior ciliary arteries) it may cause papillary edema and papillitis, which at the same time may progress to optic nerve atrophy,23,24 as in the case of our patient.
The diagnosis is predominantly clinical, although the tissue biopsy and arteriography are important tools in most patients. A positive angiogram at the time of diagnosis is more frequent in adults than in children (81% vs. 40%)25 and many times, as in this case, the biopsy findings and the images are taken over the course of the disease and the diagnosis is merely presumptive.
The treatment requires corticoids initially at high doses, whether oral or IV, and these are gradually tapered during a certain period of time as dictated by response to treatment and by the severity of the clinical manifestations; however, there are no clinical trials to guide treatment. The treatment for inducing remission uses high doses of corticoids and CYC over the first 3–6 months; upon remission, the principal maintenance therapy includes low dose glucocorticoids (0.2–0.4mg/kg/day) and oral AZA at a dose between 1 and 2mg/kg/day or 12–18 additional months. Methotrexate or leflunomide may also be used.13 Some refractory cases have been described, managed with plasmapheresis and other biologics such as rituximab, tofacitinib, infliximab and tocilizumab.14,15
Short-term treatment complications such as infections and corticoid-induced side effects, continue to be a concern in children with PAN, receiving the standard therapy6; furthermore, some of the late complications associated with treatment with CYC often include, alopecia, hematological toxicity, and to a lesser extent, urological toxicity, malignancy and infertility26 have been reported in cohorts of children with other vasculitis.27 HC is a common manifestation of CYC-induced urotoxicity in rheumatic diseases; some reviews have indicated that the oral route of CYC administration and accumulated doses may increase the risk of HC28; however, it not yet clear whether there is a cutoff dose to predict the occurrence of HC.29 The severity of HC may vary, from self-limiting mild hematuria up to a potentially lethal presentation30; the mean time for resolution of the hematuria is between 7 and 56 days. The pathological findings in the acute and subacute phases may be edema, atrophy of the vesical mucosa, ulceration and bleeding which extend throughout the bladder wall; in severe cases, necrosis and chronicity, also accompanied by stromal fibrotic changes.
In systemic vasculitis, if the genitourinary system is involved, if primarily affects the kidneys and the testicles; involvement of the urinary bladder has been described only in a few case reports and been an incidental late finding over the course of the disease.31 The clinical presentation in general is lower irritative symptoms, the radiological and cystoscopy findings are unspecific, and the diagnosis is made through histology of the bladder biopsies, which are also helpful to rule out other differential diagnoses, particularly malignancy.32,33
This clinical case is particularly striking, since it showcases the novel and unusual presentation of polyarteritis not previously reported in children, associated with the activity of the disease. In our patient, it was triggered by the insidious evolution of vasculitis and poor treatment compliance.
ConclusionIn patients with constitutional symptoms, prolonged fever and organic involvement, systemic vasculitis shall be included in the differential diagnosis. Adherence to treatment is essential for the favorable evolution of any disease. In this patient, a severe disease from the onset and poor treatment compliance, were determining factors for the development of a complication, which as far as we know, is not described in pediatric patients. The case also required intensive treatment including rituximab, a drug that has improved the prognosis of patients with severe forms of vasculitis.
Conflict of interestsThe authors have no conflict of interests to disclose and the financing of this endeavor was with the author's own resources.
Please cite this article as: Jiménez KV, Mosquera C, Margarita Quintero E. Compromiso vesical en paciente pediátrico con poliarteritis nodosa: primera descripción. Rev Colomb Reumatol. 2020;27:141–146.
