



The diagnosis of neurosarcoidosis is difficult because of its clinical and radiological polymorphism. Any part of the central nervous system may be affected, and imaging studies are usually inconclusive. The case is presented of a young patient with headache, bilateral facial paralysis, and subacute lymphocytic meningitis in whom, despite biochemical and imaging studies the origin was not identified. Using 18-PET-FDG helped to identify a hypermetabolic mediastinal adenopathy, with pathology reporting non-caseating granulomas, findings suggestive of sarcoidosis. This reflects its usefulness for the diagnosis and staging of the disease, especially in situations where there is no evidence from other imaging studies of extra-neural disease.
El diagnóstico de la neurosarcoidosis es difícil por su polimorfismo clínico y radiológico, y cualquier parte del sistema nervioso central puede estar afectado. Presentamos el caso de una paciente joven con cefalea, meningitis linfocitaria subaguda, en quien a pesar de estudios bioquímicos e imagenológicos no se identificó la etiología. La tomografía por emisión de positrones con 18 fluorodeoxiglucosa permitió la identificación de un ganglio mediastinal hipermetabólico, con informe de patología de granulomas no caseificantes, hallazgos sugestivos de sarcoidosis, reflejando la utilidad de la tomografía por emisión de positrones con 18 fluorodeoxiglucosa para el diagnóstico y estadificación de la enfermedad, especialmente en las situaciones en las que no hay evidencia mediante otros estudios imagenológicos de enfermedad extraneural.
Sarcoidosis is a systemic granulomatous disease of unknown etiology and the lung is the most commonly affected organ.1,2 The clinical presentation of neurosarcoidosis is heterogeneous, since it may present with cranial neuropathy, aseptic meningitis, cognitive impairment, seizures, or facial diplegia.3 In order to make the diagnosis, histological confirmation is required. However, in some cases, the laboratory and neuroimaging findings are not conclusive; under these circumstances, additional studies are such as 18-PET-FDG are indicated, which as in the case under discussion, allowed for the identification of hypermetabolic sites susceptible to biopsy, similar to the description for patients who are suspected to have lymphoma or cancer, with unknown primary origin.
Case description32-year old female patient presenting with global headache, bilateral facial paralysis, associated with abdominal pain over the last three weeks. Additionally, the patient claimed she had experienced weight loss (10kg in the last month), hyporexia and diaphoresis. No history of use of medications or exposure to toxic substances.
At admission the patient was dehydrated, with normal vital signs, no lesions in the oral cavity, with cervical lymphadenopathies <1cm, and normal chest and abdomen. The neurological examination reported normal mental function, normal campimetry and bilateral eye fundus, normal facial sensitivity, bilateral facial paralysis, normal lower cranial nerves, normal strength, normal reflexes with terminal neck stiffness.
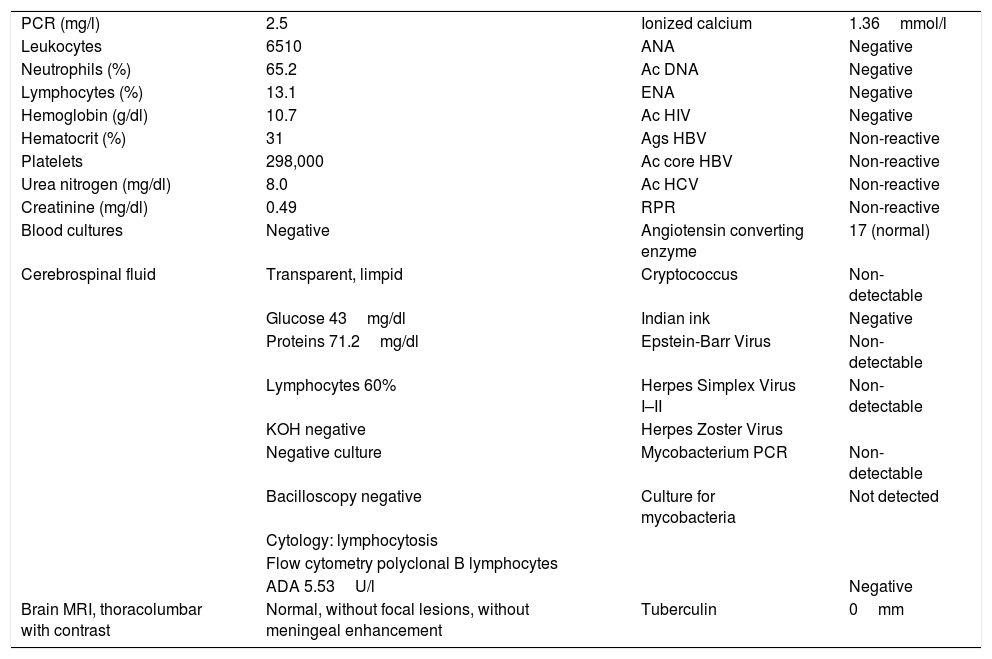
The paraclinical tests showed mild hyperkalemia, cerebrospinal fluid lymphocytic pleocytosis, hypoglycorrhachia and hyperproteinorrachia. The patient received initial empirical treatment with anti-tuberculosis 4-drug combination regime, with weak positive adenosine deaminase, negative polymerase chain reaction for Mycobacterium tuberculosis, absent MRI meningeal enhancement; negative cell clock and flow cytometry for malignancy, and negative mycobacteria culture (Table 1).
Patient's laboratory findings (blood and cerebrospinal fluid).
| PCR (mg/l) | 2.5 | Ionized calcium | 1.36mmol/l |
| Leukocytes | 6510 | ANA | Negative |
| Neutrophils (%) | 65.2 | Ac DNA | Negative |
| Lymphocytes (%) | 13.1 | ENA | Negative |
| Hemoglobin (g/dl) | 10.7 | Ac HIV | Negative |
| Hematocrit (%) | 31 | Ags HBV | Non-reactive |
| Platelets | 298,000 | Ac core HBV | Non-reactive |
| Urea nitrogen (mg/dl) | 8.0 | Ac HCV | Non-reactive |
| Creatinine (mg/dl) | 0.49 | RPR | Non-reactive |
| Blood cultures | Negative | Angiotensin converting enzyme | 17 (normal) |
| Cerebrospinal fluid | Transparent, limpid | Cryptococcus | Non-detectable |
| Glucose 43mg/dl | Indian ink | Negative | |
| Proteins 71.2mg/dl | Epstein-Barr Virus | Non-detectable | |
| Lymphocytes 60% | Herpes Simplex Virus I–II | Non-detectable | |
| KOH negative | Herpes Zoster Virus | ||
| Negative culture | Mycobacterium PCR | Non-detectable | |
| Bacilloscopy negative | Culture for mycobacteria | Not detected | |
| Cytology: lymphocytosis | |||
| Flow cytometry polyclonal B lymphocytes | |||
| ADA 5.53U/l | Negative | ||
| Brain MRI, thoracolumbar with contrast | Normal, without focal lesions, without meningeal enhancement | Tuberculin | 0mm |
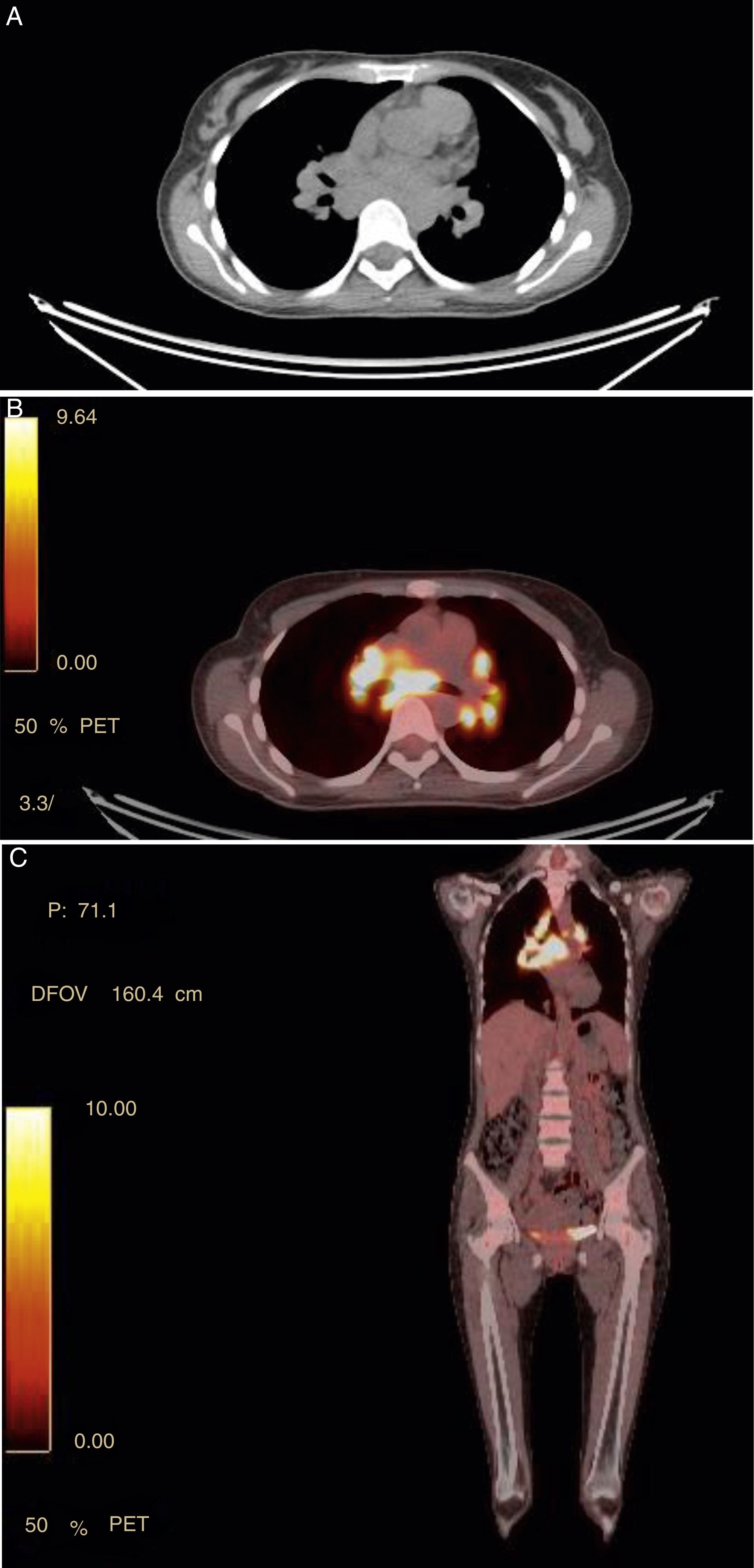
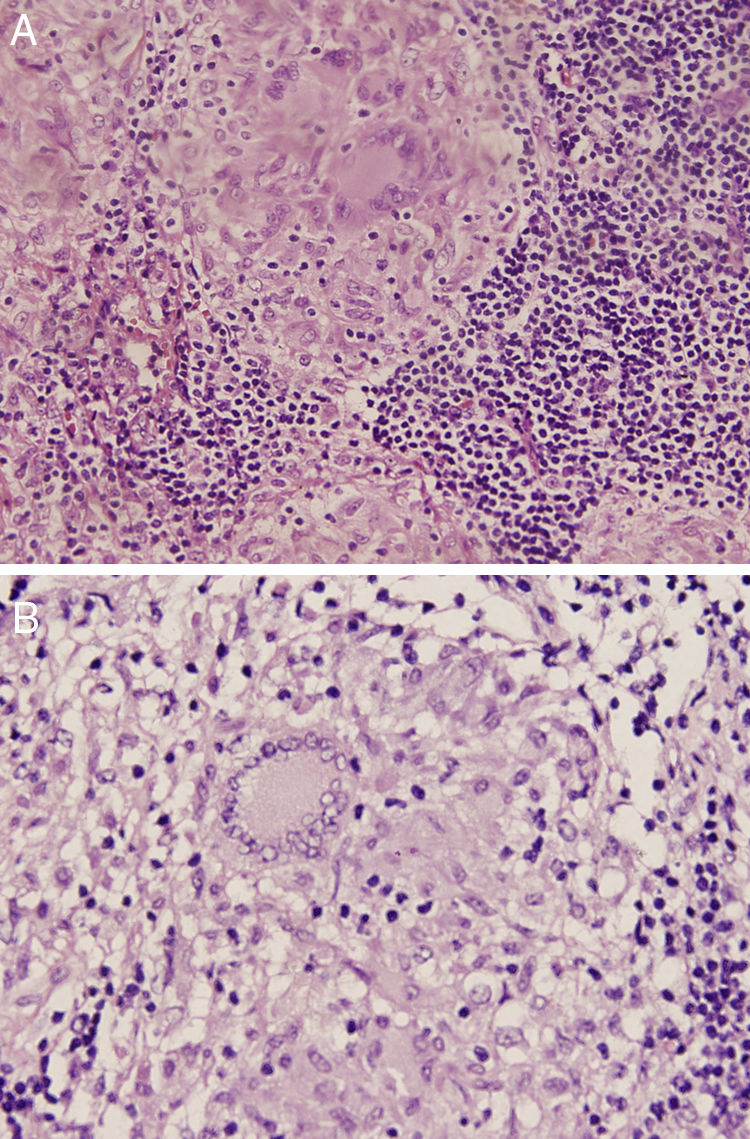
The 18-PET-FDG showed multiple hypermetabolic lymph node clusters in the cervical region, the mediastinum, and the lung hilum, suggestive of lymphoproliferative involvement (Fig. 1A–C). A mediastinal lymph node biopsy showed evidence of granulomatous lymphadenitis, multinucleated giant cells, without caseous necrosis or bacilli. Both Ziehl Neelsen and Schiff & Grocott stains were negative (Fig. 2A and B). The patient was diagnosed with neurosarcoidosis disease and received glucocorticoid therapy, achieving an optimal treatment response; over the course of clinical and imaging follow-up, the patient has remained asymptomatic.
 Panel A shows hypermetabolic lymph nodes in the upper mediastinum in FDG PET/CT. In the CT scan, the lymphadenopathies are marked with diameters between 7.7×6mm. Panel B axial image (coronal Panel C) with areas of intense uptake of FDG in mediastinal lymph nodes with SUV of 17, paratracheal upper right and left, and in bilateral hilar regions.")
(A–C) Panel A shows hypermetabolic lymph nodes in the upper mediastinum in FDG PET/CT. In the CT scan, the lymphadenopathies are marked with diameters between 7.7×6mm. Panel B axial image (coronal Panel C) with areas of intense uptake of FDG in mediastinal lymph nodes with SUV of 17, paratracheal upper right and left, and in bilateral hilar regions.
 Shows non-caseating granulomas (H–E, 5×). (B) Magnification of a sarcoid granuloma with eosinophilic epithelioid cells and multinucleated giant cells surrounded by lymphocytes.")
Sarcoidosis is a systemic granulomatous disease, with neurological complications in 5–10% of the cases (autopsies 27%).1,2 The nervous system involvement was described for the first time in 1909 by Heerfordt (“uveoparotid fever”).4,5
It may present in all ethnic groups, being most common in northern Europeans and Afro Americans, with an incidence of 15–20 and 35–80 per 100,000 inhabitants, respectively. It occurs mainly in women, with an incidence peak over the third and fifth decades of life.5
The characteristic pathological finding is non-necrotizing granuloma. The etiology is not clearly identified and exposure to mycobacteria Propionibacterium acnes, pesticides, yeasts and metals has been suggested,4 resulting in hypersensitivity in a genetically susceptible host. Other findings are elevated CD4:CD8 T-lymphocytes ratio in bronchoalveolar lavage and in the cerebrospinal fluid, chronic Th1 stimulation, with elevation of interleukin (IL)-12 and 18, interferon gamma and alpha in the bronchoalveolar fluid and granulomas.5 The granulomatous inflammation affects the leptomeninges and may invade the brain and the spinal cord through the Virchow Robins spaces.4
The most frequently used diagnostic criteria are Zajicek's; in our case, probable neurosarcoidosis, due to the neurological involvement and tissue confirmation through a mediastinal lymph node biopsy and exclusion of other etiologies.4
MRI is the initial study, with high sensitivity (90%) but low specificity, even below 33% in accordance with the parameter.5 The most frequent finding is thickening and enhancement of the meninges, particularly on the basal surface; in extensive cases, there may be axis length involvement, hydrocephalus, intraparenchymal and pituitary hypothalamic enhancement.4,6
The CSF analysis confirms the presence of inflammation and excludes infection or malignancy. It is abnormal in 70–95% of the cases, with unspecific findings such as this case: hyperproteinorrachia, hypoglycorrachia and mononuclear pleocitosis.4,7 Cytology, flow cytometry, and cultures are mandatory in all patients.4
The search for systemic disease requires serological and pulmonary evaluation; however, these are unspecific manifestations such as anemia, leucopenia, thrombocytopenia, and hypercalcemia.5 The levels of angiotensin converting enzyme in the CSF have low sensitivity (66.7%) and specificity (67.3%), and do not help in the diagnosis.8
In the absence of extraneural disease, such as the case under discussion, 18-PET-FDG and gallium-67 scintigraphy are useful to identify subclinical systemic involvement.4
The sensitivity of scintigraphy for active sarcoidosis is 97%. The characteristic finding is the Panda sign which is due to inflammation of the lacrimal glands, lesser salivary glands, and parotid glands, highly suggestive of sarcoidosis. Nevertheless, it is not specific and may be seen in Sjögren's syndrome. This study is of limited use because of its high cost and radiation-associated risks.5
18-FDG PET exhibits better sensitivity and helps in the diagnosis of neurosarcoidosis when conventional imaging is not conclusive. It also allows for assessing treatment response, with faster results due to the rapid radioactive drug uptake. Staging and identification of biopsy sites is possible, indicating the optimal hypermetabolic site for the histopathology study. The downside is the unspecific results that hinder the differentiation between sarcoidosis and lymphoma and the diagnosis requires histological confirmation.5,9
The diagnosis of neurosarcoidosis is established through a CNS biopsy. However, in suspicious cases, without systemic disease, confirmation is possible in the presence of neurological involvement and assessment of an extraneural source biopsy.4
As mentioned in this case, 18-PET-FDG allowed for the identification of multiple hypermetabolic lymph node clusters in the cervical, mediastinal, and hilar regions to guide the biopsy that confirmed the diagnosis of neurosarcoidosis, although the morphology or size were not abnormal in other images.
Most cases of neurosarcoidosis respond to corticosteroids. In mild to moderate cases, the dose of prednisone ranges from 20 to 40mg/day. In severe cases, the recommendation is to administer methylprednisolone pulses of 500–1000mg/day for 3–5 days, with maintenance doses of 0.5–1mg/kg or 60mg/day for 6–12 months. However, around 25% of the patients are resistant to corticosteroids.10
Alpha-interferon is the second line of treatment.2 Other options in refractory cases include tumor necrosis factor alpha (infliximab, adalimumab), cyclophosphamide, mycophenolate mofetil and rituximab. Radiotherapy helps in patients with pituitary dysfunction.10 The prognosis of the disease is associated with the extension and neurological involvement. 75% of the patients presenting with neurosarcoidosis will develop systemic sarcoidosis over the course of follow-up.5
ConclusionsNeurosarcoidosis is a rare disease that requires high clinical suspicion. The laboratory and imaging findings of the diagnostic approach are usually unspecific, and typically a histopathology confirmation with 18-PET-FDG-guided extraneural biopsy is required; this test is also valuable for staging and follow-up purposes during treatment, and to rule out any differential diagnoses, particularly in cases of subclinical systemic involvement.
Conflict of interestThe authors have no conflicts of interest to disclose.
Please cite this article as: Bohórquez L, Vargas DC. Utilidad de la tomografía por emisión de positrones - tomografía computada para el diagnóstico de neurosarcoidosis. A propósito de un caso. Rev Colomb Reumatol. 2018;25:211–215.
