



Clinically amyopathic dermatomyositis comprises a special group of patients within the spectrum of dermatomyositis characterized by the presence of typical skin lesions, minimal or absent muscle involvement, and increased risk of interstitial lung disease. The antibodies directed against the protein encoded by melanoma differentiation-associated gene 5 (MDA5) are present in a significant proportion of patients with clinically amyopathic dermatomyositis, who develop rapidly progressive interstitial lung disease, with high mortality and frequently complicated by the onset of spontaneous pneumomediastinum. A case is presented of an African patient with anti-MDA5 positive clinically amyopathic dermatomyositis and interstitial lung disease with tomography pattern of organizing pneumonia who developed spontaneous pneumomediastinum during its clinical course.
La dermatomiositis clínicamente amiopática comprende un grupo especial de pacientes dentro del espectro de la dermatomiositis, caracterizados por la presencia de lesiones cutáneas típicas, compromiso muscular mínimo o ausente y riesgo aumentado de enfermedad pulmonar intersticial. Los anticuerpos dirigidos contra la proteína codificada por el gen asociado con la diferenciación del melanoma 5 (MDA5), están presentes en una proporción importante de pacientes con dermatomiositis clínicamente amiopática, los cuales desarrollan enfermedad pulmonar intersticial rápidamente progresiva, con elevada mortalidad y que se complica frecuentemente con la aparición de neumomediastino espontáneo. Presentamos el caso de una paciente de origen africano con dermatomiositis clínicamente amiopática anti-MDA5 positiva y enfermedad pulmonar intersticial con patrón tomográfico de neumonía organizada, que desarrolló neumomediastino espontáneo durante su evolución.
Dermatomyositis (DM) is a systemic inflammatory disease characterized by proximal muscle weakness, myalgias and typical skin manifestations such as “heliotrope” erythema and Gottron's papules. However, some patients with DM do not develop muscle alterations (amyopathic DM), while others have mild symptoms during the course of the disease, or the muscle commitment is evidenced only by elevation of the enzymes associated with muscle damage or by myopathic alterations in the electromyogram or in the muscle biopsy (hypomyopathic DM), so they are included within the spectrum of the clinically amyopathic DM (CADM), having this group an increased risk of developing interstitial lung disease (ILD).1
Although its etiology is unknown, there is evidence that the tissue damage in DM is produced by autoimmune mechanisms, finding circulating antibodies specific for myositis such as anti-Mi2 and anti-Jo1 in 50–70% of cases.2 Recently, there have been identified antibodies directed against the protein encoded by the melanoma differentiation-associated gene 5 (MDA5), belonging to the family of Rig-I-like receptors that are related with the response to viral infections, which are present in 19–35% of the patients with CADM and are specifically associated with minimal or absent muscle involvement and rapidly progressive ILD, frequently complicated by the appearance of spontaneous pneumomediastinum (SPM).3,4 We present the case of a patient of African origin with CADM and positivity for anti-MDA5 who developed ILD and SPM during its evolution.
Case presentationAn 18-year-old black woman, native from Western Sahara, who at the age of 14 years began presenting symmetric polyarthritis, fever and alopecia. At the age of 16 years, she was transferred to Algeria, where treatment with oral prednisone (maximum dose 10mg/day) was started, developing shortly after Raynaud's phenomenon and ulcerations in the finger pads of both hands. The patient was admitted to the hospital at the age of 17 years due to dyspnea and non-productive cough, evidencing bibasilar opacities and altered spirometry with severe restrictive pattern (forced vital capacity 33% of its theoretical value), receiving intravenous empirical antibiotic therapy and being discharged with partial improvement. During this admission, positive rheumatoid factor (RF) (108IU/ml) was detected, so she was diagnosed with rheumatoid arthritis and administration of oral methotrexate (10mg/week) plus folic acid supplement (5mg/week) was started.
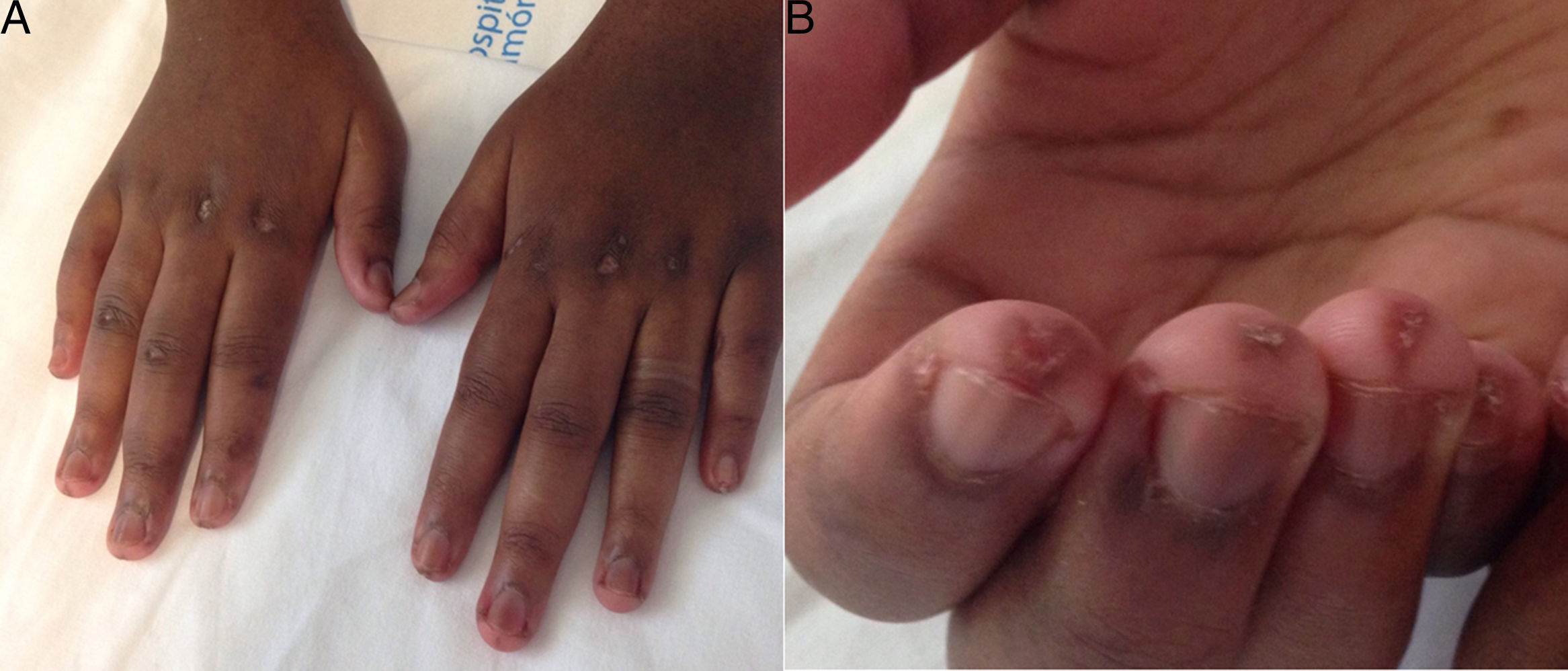
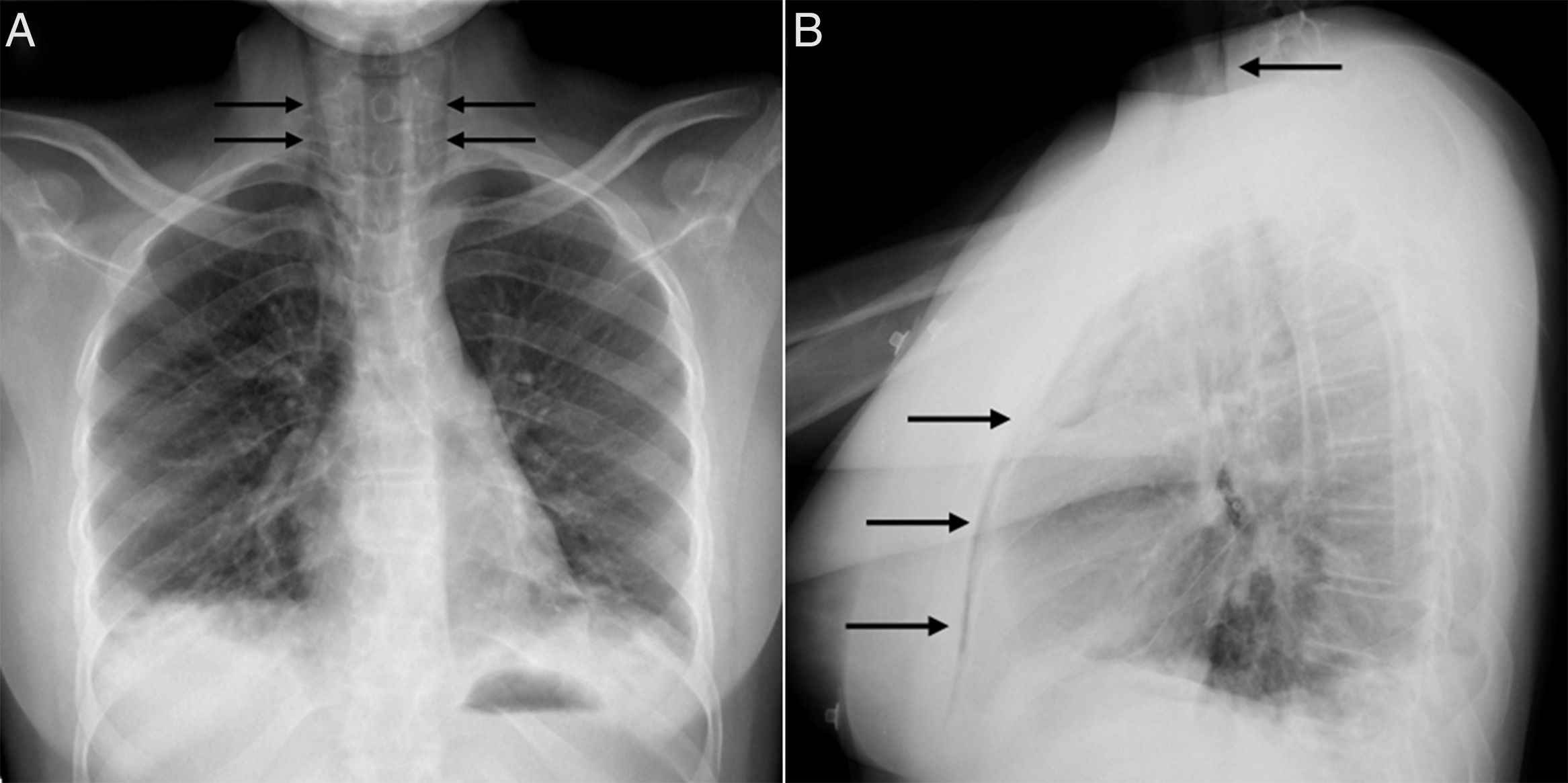
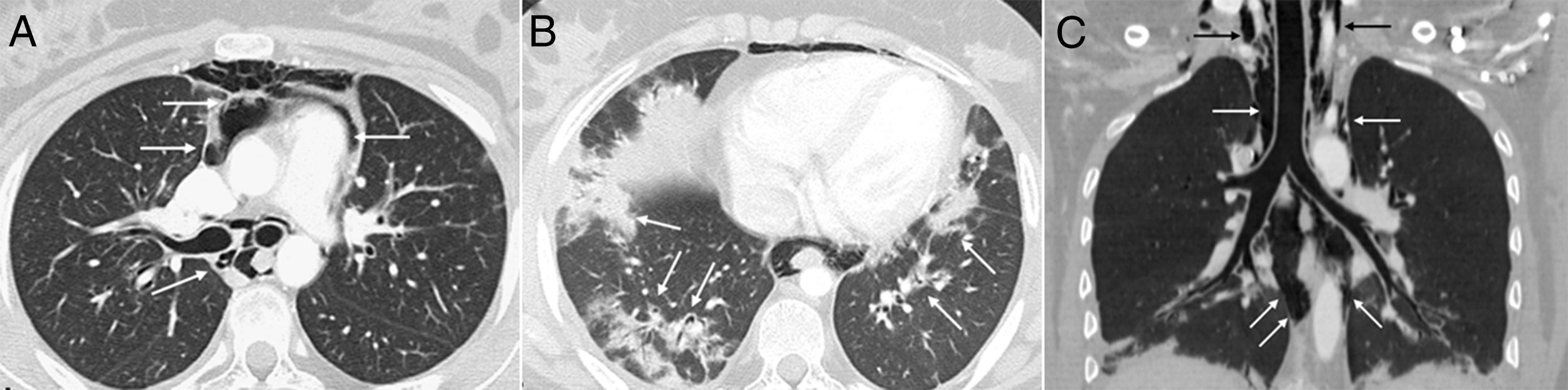
At age 18, she was transferred to Spain and admitted to our center, having at this time diffuse edema of the hands, scarring lesions in the finger pads, periungual erythema, hypopigmented macules of atrophic appearance on areas in which she had previously Gottron's papules in metacarpophalangeal and proximal interphalangeal joints (Fig. 1), indurated subcutaneous nodules in forearms and thighs, dyspnea on mild efforts, dysphagia, pyrosis and asthenia, but without muscle weakness or myalgias. The basal oxygen saturation was normal (97%) but with persistence of the restrictive pattern (forced vital capacity 34.8%), being not possible to carry out the test for diffusing capacity of the lungs for carbon monoxide (DLCO) because she mobilized an insufficient volume. The laboratory tests showed hypochromic microcytic anemia (hemoglobin 11.8g/dl, mean corpuscular volume 74.6fl, mean corpuscular hemoglobin 23.7pg), lymphopenia (750/mm3), increased acute phase reactants (erythrocyte sedimentation rate 84mm/h, C-reactive protein 17mg/l), polyclonal hypergammaglobulinemia (IgG 2800mg/dl) and increase in thyroid stimulating hormone (TSH) (5.230μIU/ml) with normal T3 and T4 levels. The rest of the biochemical study including muscle enzymes (creatine kinase 40U/l, aldolase 2U/l) was within normal ranges. From the immunological point of view she had ANA 1/80 with nucleolar pattern, positive anti-Ro52 and anti-MDA5, being negative the other antibodies studied (RF, anti-cyclic citrullinated peptide [anti-CCP], anti-DNA, anti-ENA, anti-Scl70, anti-centromere, antiphospholipid antibodies, anti-Mi2, anti-Jo1, anti-PL12, anti-PL7, anti-OJ, anti-EJ, anti-SRP, anti-Ku and anti-PM/Scl). The chest X-ray showed the bibasilar opacities and signs of SPM extending into the perithyroidal region (Fig. 2), although crepitation in subcutaneous cellular tissue was not evident in the physical examination, and for this reason it was performed a computed tomography that confirmed the presence of patchy peribronchial opacities of basilar predominance, radiologically compatible with a pattern of organizing pneumonia and an extensive pneumomendiastinum which was expanded from the thyroid region throughout the entire mediastinum dissecting vascular and muscle structures. (Fig. 3) There were no signs of pleural honeycombing, mediastinitis or pneumothorax.

, without pneumothorax or subcutaneous emphysema, and bibasilar parenchymal opacities (A) and (B).")
 (A). Another axial image demonstrates the existence of patchy peribronchial opacities in both lung bases, compatible with a pattern of organizing pneumonia (arrows) (B). An extensive pneumomediastinum that extends from the thyroid region (black arrows) throughout all the mediastinum (white arrows) is observed on the coronal reconstruction (C).")
Axial CT image of the thorax which confirms the presence of gas dissecting the structure of the mediastinum (arrows) (A). Another axial image demonstrates the existence of patchy peribronchial opacities in both lung bases, compatible with a pattern of organizing pneumonia (arrows) (B). An extensive pneumomediastinum that extends from the thyroid region (black arrows) throughout all the mediastinum (white arrows) is observed on the coronal reconstruction (C).
The patient was initially diagnosed with amyopathic DM with associated ILD and SPM, but the electromyogram demonstrated spontaneous activity suggestive of mild inflammatory myopathy, and nonspecific myopathic changes without inflammatory infiltrate were found in the muscle biopsy, therefore, it corresponded actually to hypomyopathic DM. The study was completed with capillaroscopy which showed isolated megacapillaries with preserved capillary density, esophageal manometry with weak contractions at the level of the lower half of the esophagus, and biopsy of the subcutaneous nodules with findings compatible with nonspecific panniculitis. Administration of prednisone 1mg/kg/day was initiated with rapid improvement of dyspnea, cutaneous lesions and asthenia. Methotrexate was discontinued and azathioprine 100mg/day was added. In the evaluation 2 months after discharge the patient was stable, with mild dyspnea and arthralgias without signs of synovitis. The chest X-ray showed an important reduction of the pulmonary infiltrates and disappearance of the signs of SPM. The patient abandoned the follow-up due to change of domicile.
DiscussionAnti-MDA5 antibodies (formerly called anti-CADM140) were described in 2005 by Sato et al., in Japanese patients with CADM and rapidly progressive ILD.5 Further studies have found these antibodies in other populations and expanded their spectrum of clinical manifestations, being characteristic the presence of cutaneous and oral ulcerations, painful palmar papules/pustules, periungual erythema, erythema in elbows/knees (Gottron's sign), diffuse alopecia, panniculitis and arthralgias/arthritis.3,4,6 Due to these characteristics, as well as to the scarce muscle involvement, the preponderance of the ILD and the infrequent association with neoplasms, it has been proposed to call this picture “dermatopulmonary syndrome associated with MDA5 antibodies” to differentiate it from the classical DM,6 although these antibodies are not exclusive to CADM and they have been also described in the classical and juvenile forms of the DM.7 The ability of anti-MDA5 antibodies to identify patients with DM and the risk of developing rapidly progressive ILD is high, with a sensitivity of 77% and a specificity of 86%.7 The racial factor can influence the clinical expressivity in patients with anti-MDA5. Approximately half of patients coming from East Asia (Japan, China and Korea) have rapidly progressive ILD and it generally corresponds to CADM,8 while in Caucasian populations the severity of the ILD may be lower and the frequency of clinical myositis higher.9 CADM associated with anti-MDA5 has been described in African American patients,9,10 but according to the literature review we have conducted, this is the first case in a woman of African origin.
In our case, the diagnosis of RA was initially proposed because of the presence of arthritis and positive RF, a fact that is not uncommon in patients with positive anti-MDA5, since a significant proportion (65.5–81.8%) develop symmetric polyarthritis with involvement of the small joints of the hands and morning stiffness that is indistinguishable from RA, some of whom may show positivity for RF or anti-CCP and, more rarely, erosions.3,9,11 As well, is not uncommon the development of arthritis associated with “mechanic's hands”, Raynaud's phenomenon or fever in patients with positive anti-MDA5, which can also give rise to the suspicion of an antisynthetase syndrome (ASS), being recommended the determination of these antibodies when the antisynthetase antibodies are negative.9 Our patient also had anti-Ro52 antibodies, which have been described as co-stimulators in the antisynthetase syndrome, increasing the severity of ILD in these cases.12,13 This has also been reported in patients with anti-MDA5 positive CADM, finding the simultaneous presence of anti-Ro52 in 19–50% of cases.3,9,14 The coexistence of both antibodies probably has implications for the pathogenesis of the ILD in this subgroup of DM. MDA5 and Ro52 (TRIM21) are cytoplasmic proteins whose expression is induced by type I interferons, therefore, it has been suggested that the interactions of both molecules could lead to the formation of molecular complexes with increased immunogenicity.9
A peculiar characteristic of the ILD associated with DM is the frequent occurrence of SPM, with an incidence ranging between 2.2% and 8.3%, and more than half of these cases corresponding to CADM.15,16 The presence of ILD, cutaneous vasculopathy, the use of systemic glucocorticoids, a young patient and normal serum levels of muscle enzymes have been described as risk factors for developing SPM,15 all of them present in our case. The presence of anti-MDA5 also seems to be a predictive factor for the development of SPM in these patients, as demonstrated by the study conducted by Koga et al., which included 79 patients with DM (58 classical and 21 amyopathic), finding that the presence of mediastinal emphysema was significantly greater in patients with positive anti-MDA5 (35% vs. 2%, p=2.1×10−5).4 The mechanism by which the SPM develops in DM is not clearly established, but it is speculated that it could be the result of the rupture of subpleural bullae or cysts secondary to the interstitial fibrosis and the increased intra-alveolar pressure. It has also been described a weakening of the alveolar walls due to the treatment with glucocorticoids.17,18
Regarding the tomographic patterns of ILD found in patients with positive anti-MDA5, the most frequent is that with basilar consolidation/ground glass areas (50%), followed by ground glass areas with a random distribution (33%), while the basilar reticular and peribronchovascular consolidation patterns suggestive of organizing pneumonia are usually infrequent.19 The histopathology of ILD associated with anti-MDA5 positive CADM has not been systematically evaluated, but in the case reports with lung biopsy or autopsy, the findings were compatible with diffuse alveolar damage, the fibrotic variant of nonspecific interstitial pneumonia and the usual interstitial pneumonia.6,14,18-24
The majority of patients with ILD associated with DM have a favorable response to treatment with high-dose glucocorticoids and immunosuppressants. However, those with CADM and rapidly progressive ILD are resistant to multiple treatments and have a poor prognosis, with a mortality of 41% in patients with positive anti-MDA5, so it is recommended to treat them early and aggressively with combinations of drugs including glucocorticoids, calcineurin inhibitors, mycophenolate mofetil, intravenous immunoglobulins, cyclophosphamide and rituximab.4,8,25 The presence of SPM also influences prognosis, finding a mortality rate of 34% in patients with DM who have this complication, with 25% of them dying during the first month due to respiratory distress.16 There have been published 2 cases of rapidly progressive ILD in CADM associated with anti-MDA5 that were refractory to treatment with pulses of methylprednisolone and calcineurin inhibitors and finally underwent hemoperfusion with polymyxin B, a technique that was originally developed to remove the endotoxin from the circulation in patients with sepsis, which has demonstrated to be effective in patients with respiratory distress. One of them showed dramatic improvement and reduction in the levels of anti-MDA5 antibodies,26 but the other case (who presented SPM and subcutaneous emphysema as complications) only had a slight transient improvement and required plasmapheresis and intravenous immunoglobulins.27
In conclusion, the anti-MDA5 antibodies identify a special group of patients with DM who have a tendency to present amyopathic/hypomyopathic forms, with a spectrum of mucocutaneous and articular manifestations and an increased risk to develop ILD, which can follow a rapidly progressive course and be complicated by the appearance of SPM.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare they do not have any conflict of interest.
Please cite this article as: Sifuentes-Giraldo WA, García-Villanueva MJ, Gorospe L. Neumomediastino espontáneo como complicación de enfermedad pulmonar intersticial, asociada con dermatomiositis clínicamente amiopática y anticuerpos anti-MDA5 positivos. Rev Colomb Reumatol. 2017;24:259–264.
