




Behçet's disease is a clinical autoinflammatory disorder of unknown etiology, usually with systemic manifestations, and a pattern of exacerbation-remission, often associated with delayed diagnosis.
The diagnosis of this disease is complex. This article discusses four cases of patients with Behçet's disease, who during the clinical approach were considered to have other autoimmune diseases. A comprehensive review of the medical history, the development of oral and genital ulcers, in addition to the study of major histocompatibility complex typing (HLA), enabled the diagnosis of Behçet's disease.
La enfermedad de Behçet (EB) es una entidad clínica autoinflamatoria, de etiología desconocida, generalmente con compromiso sistémico, con un patrón de exacerbación y remisión frecuente que se asocia a retraso en el diagnóstico.
El diagnóstico de esta enfermedad es complejo, por esta razón presentamos 4 casos de pacientes con EB, que durante el abordaje clínico fueron consideradas otras enfermedades de naturaleza autoinmune. La revisión integrada de la historia clínica, la aparición de úlceras orales y genitales, así como el estudio de tipificación del complejo mayor de histocompatibilidad (HLA) permitieron diagnosticar la EB.
Behçet’s disease (BD) also referred to as Behçet’s syndrome, is a chronic autoinflammatory clinical condition of unknown etiology, classified in the group of vasculitis affecting blood vessels of variable size, not associated with ANCA.1 It is characterized by systemic involvement, affecting mostly the mucocutaneous tissue, the joints and the eyes.2 The mean age of onset of the disease is between 20 and 40 years old, affecting both genders equally.3 BD exhibits a variable clinical course in most patients, with a pattern of exacerbation and remission,4 which is usually associated with delayed diagnosis; in general, based on the reports of the various international cohorts, it takes more than 5 years to make the diagnosis.5
The prevalence of BD varies with the geographical area, but it is more frequent in the countries of the Silk Road, from the Mediterranean to the Far East Asian countries.6 A genetic predisposition associated with HLA has been identified, that facilitates its presentation and diagnosis.7 HLA-B51 stands out, for which more than 200 polymorphisms have been described.8–10 The highest frequency has been reported in Turkey, with approximately 420 patients per 100,000 inhabitants, followed by Israel, Japan, Korea and China.11 In Colombia, the prevalence of BD is 1.10 patients per 100,000 inhabitants.12
The diversity of the clinical spectrum, the asynchronous nature of the clinical manifestations, the differences in meeting all of the classification criteria, and the clinical pattern of exacerbation and remission characteristic of this condition, hinders the diagnosis of BD,13 and turns it into a clinical challenge giving rise to poorly effective therapeutic approaches.14 Since 2010, BD is considered an orphan disease in Colombia, of particular interest to the local rheumatology practice. The objective of these case series is to contribute to our understanding of the behavior of BD in our context, sharing relevant clinical data for a timelier diagnosis by the clinician.
Materials and methodsA descriptive, retrospective study of a case series including 4 non-consecutive outpatients with a diagnosis of BD was conducted. The patients were treated at the rheumatology outpatient clinic of Hospital Militar Central de Bogotá, Colombia. The medical records were reviewed and the information was collected in a preestablished form. The socio-demographic and disease-associated variables collected were: age, gender, initial symptom, organ involvement relating to BD, immunosuppressive therapy prescribed, differential diagnoses, laboratory tests (radiological images, autoantibodies profile, HLA genetic study and acute phase reactants) and time in years from the onset of symptoms and the diagnosis of BD. The results were analyzed with descriptive statistics, using Microsoft Excel 2013 spread sheet.
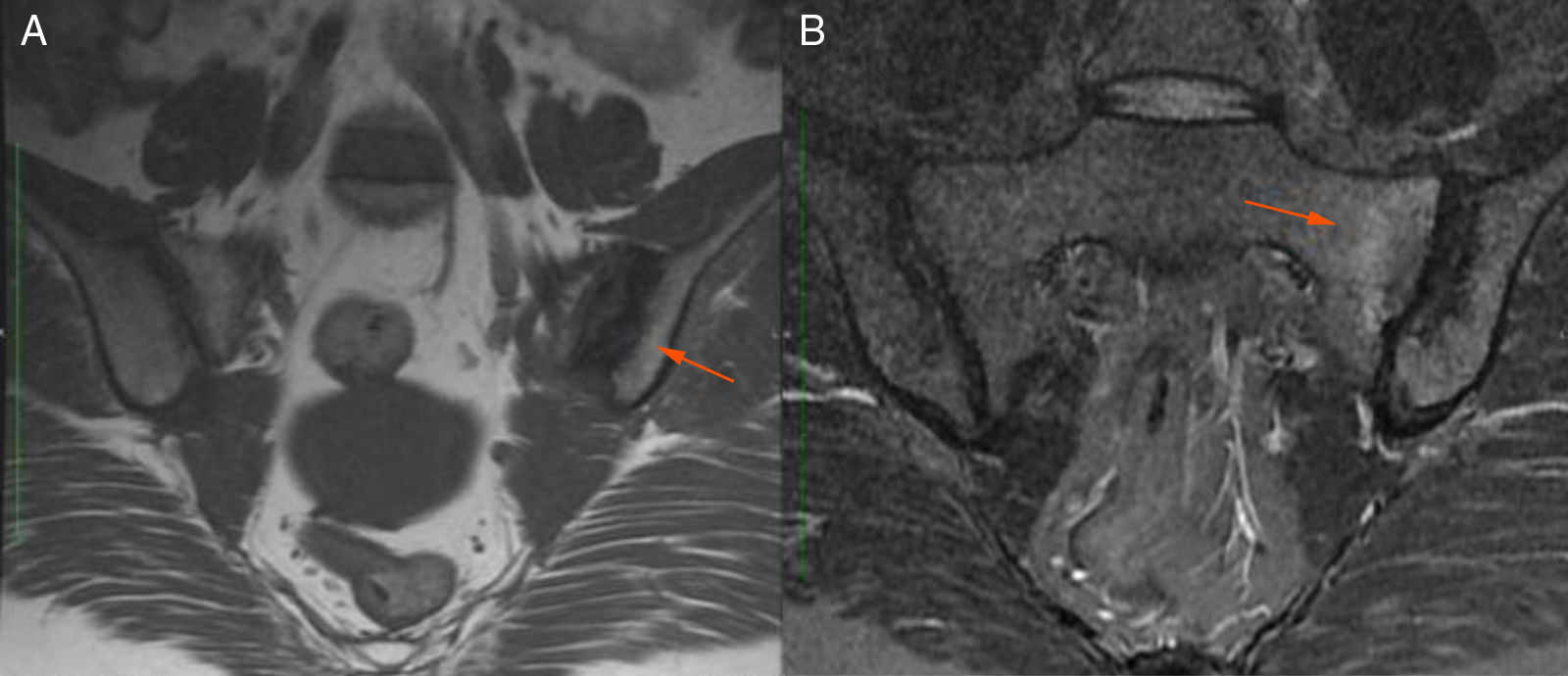
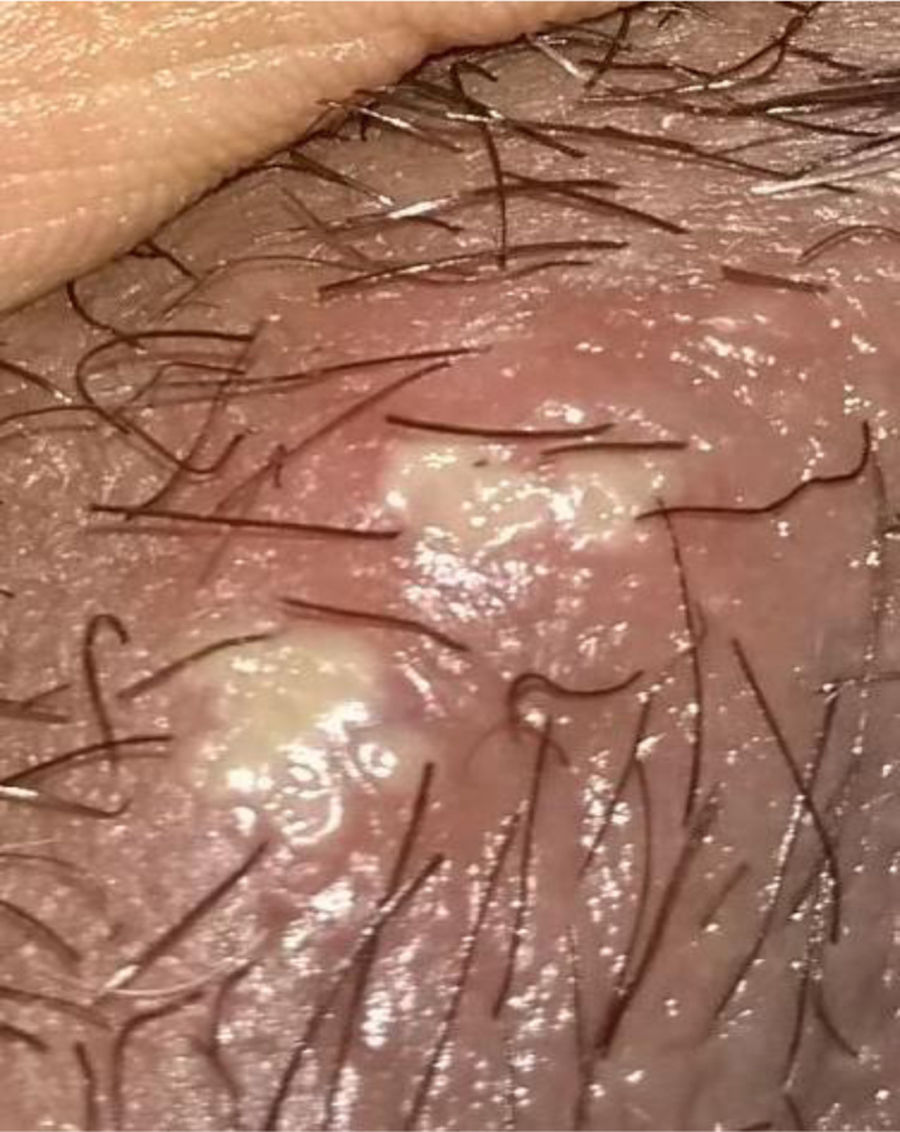
Description of the case seriesCase 137-year old female, professional nurse, who presented the initial symptoms at 23 years, including fatigue, polyarthritis, left gluteal pain radiating to the ipsilateral hip, malar erythema, photosensitivity and hair loss. The patient did not mention any personal or family history, neither any additional symptoms besides the assessment by systems. Based on the description of the condition, the suspicious diagnosis was SLE. The immune profile reported: ANA 1:160 homogeneous pattern, negative anti-DNA, normal C3-C4 and negative rheumatoid factor (RF); the acute phase reactants were initially negative and then remained slightly elevated. Combined therapy with high dose systemic glucocorticoids (SGC) and immunosuppressors (methotrexate, azathioprine and hydroxychloroquine) was prescribed, but the patient did not improve. Four years later, the patient presented with recurrent painful mouth ulcers, myalgias, and headache with alarm signs. The diagnostic approach included a brain MRI that showed increased bilateral periorbital tissue and predominant left eye exophthalmos. The lumbar puncture reported an opening pressure of 40cm of H20 and normal cerebrospinal fluid (CSF). The patient was diagnosed with idiopathic intracranial pressure (IIP) which improved with CSF drainage. Over the next 10 years, the patient experienced exacerbation and remission episodes of the mucocutaneous and joint symptoms. Due to persistent gluteal pain, an MRI of the sacroiliac joints was performed, showing subchondral bone edema and erosion of the left sacroiliac joint, compatible with sacroiliitis (Fig. 1). At 37 years old, the patient presented with multiple episodes of genital ulcers (Fig. 2); the suspicious diagnosis was sexually transmitted disease for which serological studies were conducted, including HIV, VDRL and IgM/IgG for herpes virus with negative results. One year later, the patient was assessed by rheumatology. Considering the history of oral and genital ulcers, sacroiliitis, arthritis, IIP and constitutional symptoms, BD was suspected. A pathergy test was administered and it was positive; the HLA typing reported: HLA-A*01,*24, HLA-B*35,*38 and HLA-DR*04;*13. Based on these information, the diagnosis of BD was confirmed, after 15 years of disease progression and symptoms. Initial therapy with colchicine 0.5mg every 8h and diclofenac 50mg every 12h was administered for 3 months, without any improvement; adalimumab treatment was initiated with a favorable clinical response.
 T1 sequence. Visible erosions in the left sacroiliac joint (arrow). B) STIR sequence. Visible subchondral bone edema (arrow) in the upper and lower quadrants of the left sacrum.")

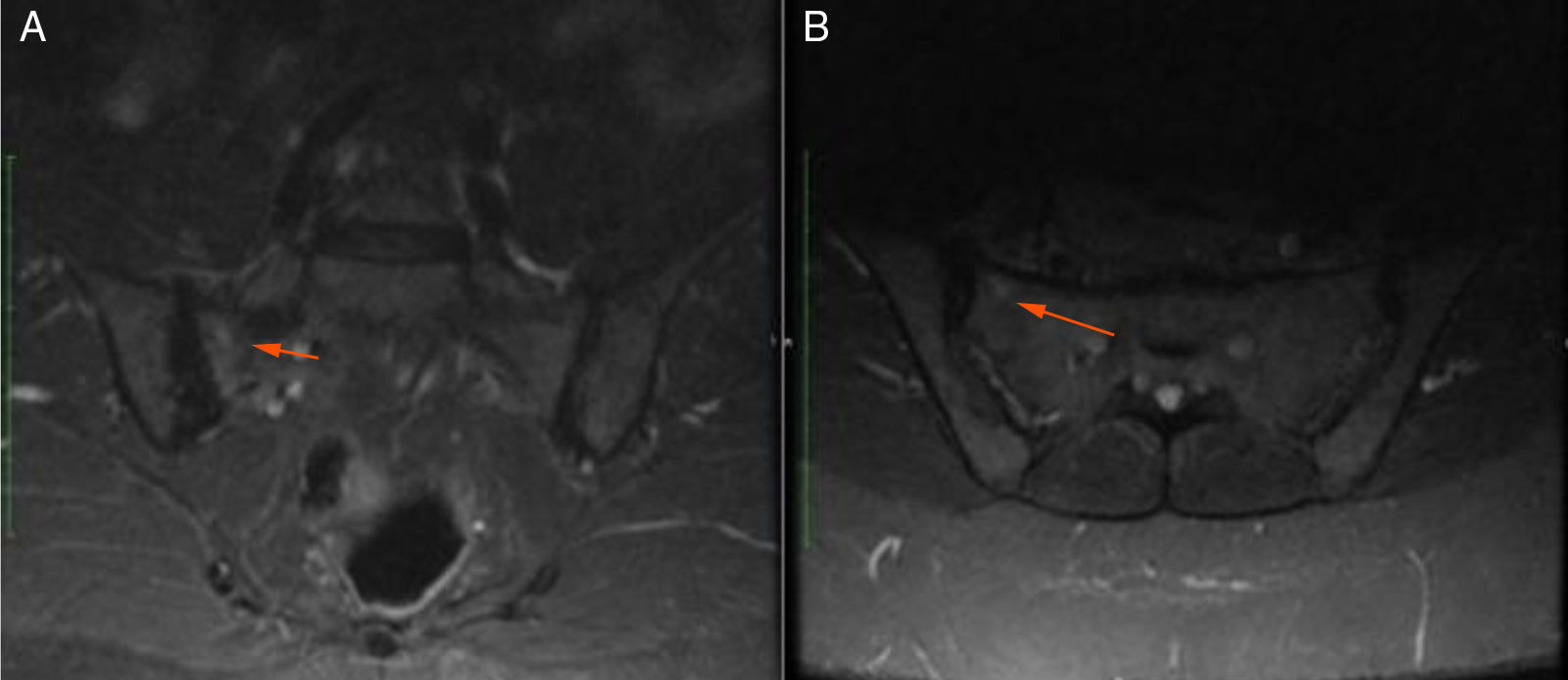
48-year old female, professional nurse. She experienced initial symptoms at age 30, including fatigue, headache and polyarthritis. The only medical history is hypertension and no additional symptoms were reported. The patient was assessed by rheumatology and treatment for suspicious autoimmune arthritis was initiated. An immune profile was conducted including: ANA, anti-DNA, ENA, anti CCP and RF, all reported negative; acute phase reactants ESR and PCR were negative. In view of the negative test results, her symptoms were considered to be secondary to fibromyalgia, and neuromodulator therapy was prescribed (gabapentin, pregabalin, amitriptyline) but her symptoms did not improve. At 33 years of age, the patient presented for the first-time recurrent mouth sores, and was diagnosed with infectious gingivostomatitis. Over the next 15 years, the articular and mucocutaneous symptoms presented intermittently, and at 48 years old, the patient presented inflammatory lumbar pain. In view of the new symptom, rheumatology adopted a new diagnostic approach with studies including HLA typing that reported negative HLA-B27 and positive HLA-B51; the MRI of the sacroiliac joints showed subchondral bone edema in the right sacroiliac joint, compatible with sacroiliitis (Fig. 3). Based on the patient’s history of polyarthritis, sacroiliitis, recurrent mouth sores and HLA-B51 positivity, after 18 years of the onset of symptoms, the diagnosis of BD was confirmed. The pathergy test was negative. Currently the patient is being treated with NSAIDs, with partial improvement and is in the process of initiating biologic therapy.
Case 3 A) Coronal B) Axial.")
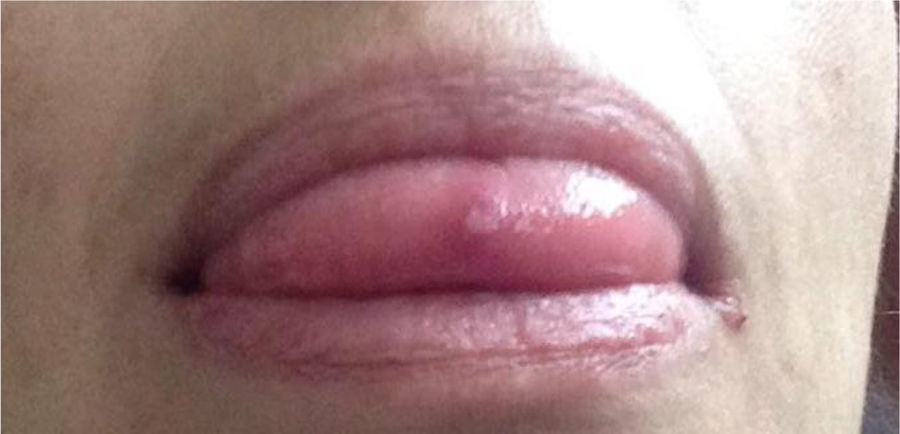
49-year old female, works in advertising, with unremarkable personal history. When she was 10 years old, the patient experienced polyarticular pain, intermittent febrile episodes, and recurrent mouth and genital sores. She was assessed by gynecology and dermatology, but no final diagnosis was made. When she was 20 years old, the symptoms spontaneously abated and she remained asymptomatic for 26 years. At 46 years old, the patient experienced an infectious episode, probably form Zika virus, and the arthritis, fatigue, and mouth and genital sores relapsed (Fig. 4). The joint disease compromised her carpal and metacarpophalangeal joints, elbows, shoulders, low back and knees. The patient was referred to the rheumatology department where she was studied for a differential diagnosis of autoimmune SLE disease vs. inflammatory arthritis. The tests performed included: anti-DNA, ANA, ENA, RF and negative CCP; RNMSI were compatible with bilateral sacroiliitis; the HLA typing reported HLA-A*02;*24, HLA-B*35;*51, HLA-DR*04;*16. Based on her clinical presentation and the above laboratory tests, after 39 years of the onset of symptoms, the diagnosis of BD was confirmed. She was prescribed glucocorticoids (deflazacort 6mg/day) and colchicine 0.5mg tablets every 8h for 12 months, with failed clinical response. The decision was made to start biologic therapy and the patient initially received etanercept, which had to be discontinued due to a severe allergic skin reaction; she was switched to adalimumab and has been receiving treatment for 16 months, with favorable clinical response.
Case 4
30-year old female doctor, who presented her initial symptoms when she was 16 years old. Her personal and family history was unremarkable and the systems assessment was negative. The initial clinical presentation was characterized by fatigue, arthritis of the metacarpophalangeal, proximal interphalangeal and carpal joints, alopecia areata, herpes-like mouth sores and involuntary weight loss. The symptoms presented intermittently and asynchronously. The only abnormal paraclinical finding was persistent elevated ESR. The immune profile included: ANA, ENA and RF which was reported negative on several occasions. The patient was treated with cycles of NSAIDs with partial improvement but no definitive diagnosis was made. The patient was assessed by internal medicine when she was 20 years old, and base on her clinical presentation and the above laboratory tests, she was diagnosed with undifferentiated connective tissue disease. She was prescribed glucocorticoids and hydroxychloroquine, with mild improvement of symptoms. Four years later the patient developed left acute amaurosis which in the opinion of the ophthalmologist was secondary to post-chiasmal optic neuritis. She was assessed for the first time by rheumatology and still with a presumptive diagnosis of undifferentiated connective tissue disease, high dose GCS were prescribed, with partial recovery of her vision. Once again, laboratory tests were conducted because of suspicious autoimmune disease, including: ANA, ENA, anti-DNA and a profile for antiphospholipid antibodies syndrome that were reported negative. High dose GCS therapy was continued; however, the hands, knees and ankles arthritis showed no clinical improvement, and there were persistent recurrent mouth sores. Two years later, the patient experienced a second episode of left optic neuritis with similar characteristics as the previous episode, requiring once again treatment with methylprednisolone pulses. One year later, painful genital ulcers in the labia majora and the vulva. Sexually transmitted diseases were ruled out and the therapeutic regimen remained unchanged. Considering her history of polyarthritis, mouth sores, constitutional symptoms, optic neuritis and negative immune profile, rheumatology confirmed the diagnosis of BD after 14 years of the onset of symptoms. Azathioprine and colchicine were added to her treatment for 12 months, with partial remission of symptoms. Then the clinical presentation deteriorated and was prescribed adalimumab. The patient has been on adalimumab for 17 months, with favorable clinical response.
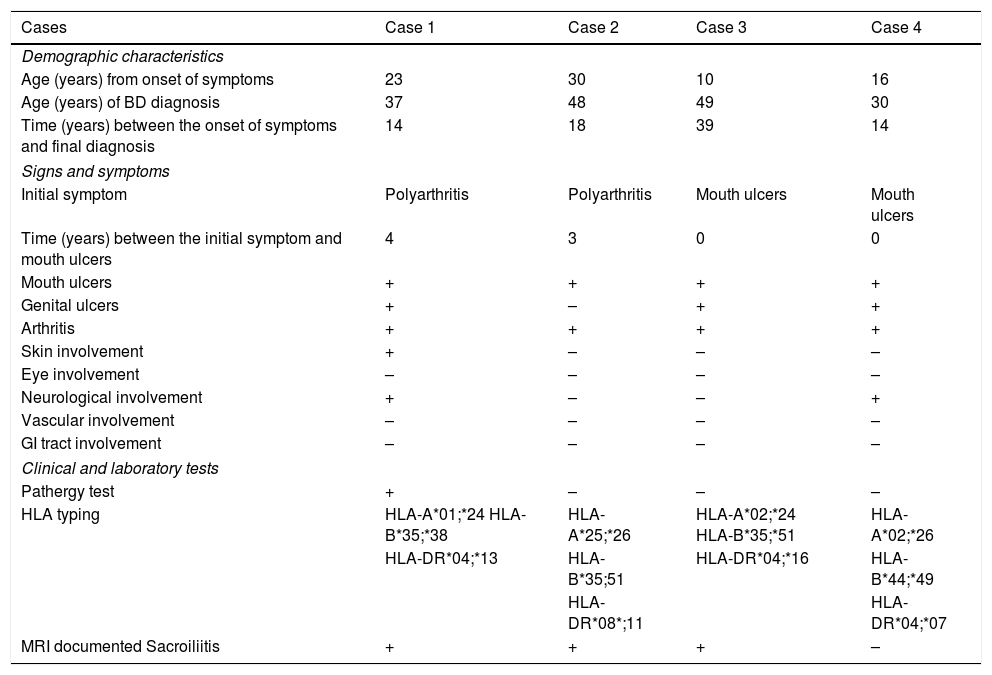
Table 1 shows the demographic characteristics and laboratory findings of the 4 patients discussed.
Description of the demographic characteristics and clinical and laboratory findings.
| Cases | Case 1 | Case 2 | Case 3 | Case 4 |
|---|---|---|---|---|
| Demographic characteristics | ||||
| Age (years) from onset of symptoms | 23 | 30 | 10 | 16 |
| Age (years) of BD diagnosis | 37 | 48 | 49 | 30 |
| Time (years) between the onset of symptoms and final diagnosis | 14 | 18 | 39 | 14 |
| Signs and symptoms | ||||
| Initial symptom | Polyarthritis | Polyarthritis | Mouth ulcers | Mouth ulcers |
| Time (years) between the initial symptom and mouth ulcers | 4 | 3 | 0 | 0 |
| Mouth ulcers | + | + | + | + |
| Genital ulcers | + | – | + | + |
| Arthritis | + | + | + | + |
| Skin involvement | + | – | – | – |
| Eye involvement | – | – | – | – |
| Neurological involvement | + | – | – | + |
| Vascular involvement | – | – | – | – |
| GI tract involvement | – | – | – | – |
| Clinical and laboratory tests | ||||
| Pathergy test | + | – | – | – |
| HLA typing | HLA-A*01;*24 HLA-B*35;*38 | HLA-A*25;*26 | HLA-A*02;*24 HLA-B*35;*51 | HLA-A*02;*26 |
| HLA-DR*04;*13 | HLA-B*35;51 | HLA-DR*04;*16 | HLA-B*44;*49 | |
| HLA-DR*08*;11 | HLA-DR*04;*07 | |||
| MRI documented Sacroiliitis | + | + | + | – |
A series of 4 cases of patients with BD is discussed, with the most salient symptoms including: arthritis, mouth and genital ulcers, and constitutional symptoms. All of the patients were females, and the mean age at diagnosis was 40.7 years. These findings are consistent with the study on the prevalence of BD in Colombia, which identified 523 cases with a female: male ratio of 2.15:1 and a higher prevalence of the disease between 45 and 49 years of age. In terms of the gender distribution, most worldwide studies show similar frequencies in both genders, but in Latin American countries (Brazil and Colombia)12,15 there is an overwhelming majority of females affected (68% in Colombia). This phenomenon may be associated to the ethnicity of the Latin American population, and further studies are needed to prove this theory.
In 3 of the 4 patients, the initial suspicious diagnosis was SLE, and other conditions such as RA and Spondyloarthritis (SpA) were considered. The diversity of the clinical spectrum, the asynchronous presentation of the manifestations and the low prevalence of BD hindered the clinical approach, with the consequence of 21.5 years of average delay in making the diagnosis, and in every case, the diagnosis was made by a rheumatologist.
In populations with a higher prevalence of BD, the diagnostic delay reported is 5.32±6.3 and 4.54±4.07 years, in a European multicenter study and in a Greek population, respectively.5,16 In Colombia, Toro-Giraldo et al. described in 2009 a series of 20 cases of BD and found that the mean time between the initial manifestation and meeting of the diagnostic criteria was 4.45±4.19 years.17 In this case series, the diagnostic delay is significantly longer than the reports from other national and international cohorts, evidencing the lack of clinical suspicion of this condition. The factors that could have contributed to this situation are: 1) long remission of symptoms; 2) late assessment by rheumatology; and 3) extended time between the initial presentation of symptoms and the development of mouth ulcers.
Epidemiological considerationsIn Colombia, a study on the prevalence of rheumatic diseases based on the registry of rheumatic diseases of the Integral System of Information on Social Protection (SISPRO) between 2012 and 2016, estimated a prevalence of 0.001% (estimated prevalence of 1.10/100,000 inhabitants and 1.49/100,000 inhabitants in people over 18 years old).12
There are no studies on the prevalence of BD in Latin America; however, there is a significant difference between the local prevalence and the Silk Road countries where it is significantly higher: Turkey (20–420/100,000), Israel (120/100,000), Iraq (17/100,000) and Japan (15/100,000); and to a lesser extent, northern Europe (Germany 2.26/100,000), United States (0.3–6.6/100,000) and England (0.64/100,000).4 This epidemiological behavior es related to the existence of a genetic predisposition that facilitates the development and diagnosis of BD.7
Clinical evolutionFrom the clinical perspective, the heterogeneity of the condition is striking. The most frequent manifestations are: mouth sores (90-100%), genital sores (85%), skin involvement (85%), eye involvement (50-70%), joints (30-50%), gastrointestinal (11%), CNS (5-10%) and vascular (4%).11 In the Colombian population, the description of case series document similar findings, with the most common clinical manifestations in all cases being mucocutaneous, eye and joint involvement.18
The most clinically relevant differential diagnoses for BD are: SLE,14,19 RA and SpA.20 To a large extent, this is due to the fact that these pathologies share similar articular, mucocutaneous, and ocular symptoms, in addition to being more prevalent than BD in our environment.21
Mucocutaneous involvement. A classical clinical characteristic of BD are mouth and genital ulcers or sores, present in almost 100% of the cases as oral manifestations and 85% for genital presentation; this becomes a distinctive clinical trait of the condition and therefore, represent 2 of the major diagnostic criteria, respectively.6 Mouth ulcers are the initial symptom in 76% of the cases and must relapse at least 3 times in one year to be considered a manifestation of BD.4,22 Classically, these sores are described as erythematous raised lesions that turn into ulcers after one or two days. Mouth ulcers are localized in the oral mucosa, the tongue, the lips and gums (less frequently in the palate, the posterior pharynx, and the tonsils), while the genital sores are localized in the scrotum, the penis, the perianal region or the vulva.23,24
Clinically, the ulcers are described as small lesions (<10mm), round or oval shape, superficial and covered by a white-yellowish pseudo-membrane.18,25 Genital ulcers are macroscopically similar to the mouth sores, but in general are larger, deep and painful, and take longer to go away than the oral sores.26 Other clinically relevant skin manifestations different from oral/genital ulcers in BD are: papulopustular lesions or pseudo folliculitis (acne like), erythema nodosum, superficial thrombophlebitis, skin ulcers or nodules, cellulite-like lesions, and pyoderma gangrenosum, and varied vasculitis-type skin lesions.25
Of the cases discussed, all of the 4 patients developed oral sores and 3 of the 4 genital ulcers. None of the patients developed skin lesions different from ulcers. In contrast to SLE and SpA, BD ulcers are painful, do not develop scars and abate within one to two weeks. While SLE and SpA are clinically relevant symptoms, the frequency of presentation is significantly lower than in BD (28%-52% for SLE and 14.5% for SpA, respectively).27–29
Articular involvement. In BD, the classical involvement is non-erosive, inflammatory oligoarticular, symmetrical or asymmetrical, intermittent and compromising the large joints (knees, ankles, elbows, and wrists).11 Less frequently, findings of enthesitis, sacroiliitis, erosive damage with loss of cartilage, pannus formation and osteonecrosis with multiple osteolytic reversible lesions are reported.4 In the cases herein discussed, 3 of the 4 patients had documented sacroiliitis, a finding consistent with the reports from other cohorts.30 In contrast, in SLE and RA the clinic is polyarticular, peripheral, classically of the small joints, symmetrical and additionally characterized by positive specific antibodies for each presentation (ANA, anti-DNA, anti-Sm, anti-nucleosome and anti-C1q for SLE and RF and anti-CCP for RA).31 BD is different from SpA from the articular point of view, because in SpA pain is typically axial or axial/peripheral, and there is a clearly defined genetic association with HLA-B27 and HLA-B15.32
Ocular involvement. Present in 50-70% of the cases and represents a major diagnostic criterion for this condition. It may compromise any ocular structure, with a prevalence of the anterior eye chamber which may lead to irreversible loss of vision.33 Bilateral involvement in 90% of the cases is more frequent in males. The clinical manifestations are ocular pain, blurred vision, and complete or partial vision loss. The typical ophthalmological pathology associated with BD is uveitis and less frequently retinal vasculitis, iridocyclitis, vitritis and papillitis.34 In other autoimmune diseases such as SLE and RA, eye involvement is less common (30% and 15-20%, respectively) and the most frequent ophthalmological pathology is Keratoconjunctivitis sicca for both SLE and RA.35–37 In the case of SpA, the prevalence of eye involvement is higher than in SLE and RA, with the SpA subtype of anchylosing spondylitis (ASp) being the one with a higher frequency of eye involvement (anterior uveitis in 25%).38 In the cases discussed, none of the patients presented ocular involvement. The optic neuritis in patient 4 is considered a neurological manifestation.
Other systems compromised in BD. BD is a multisystemic vasculitis and hence may potentially affect any system in the body. The most clinically relevant in terms of morbidity and mortality are: neurological compromise (5-10%), vascular (2-37%) and gastrointestinal (3-26%).6,13,39
Neurological involvement. May affect the central nervous system (CNS) and the peripheral nervous system (rarely). The CNS involvement may be classified into parenchymatous (80%) and non-parenchymatous (20%).40,41 The former includes: extensive brain stem/basal ganglion lesions (the isolated brain stem atrophy is almost pathognomonic of BD), hemispheric manifestations, spinal cord injuries, and aseptic meningoencephalitis. Clinically it manifests with pyramidal signs, hemiparesis, behavioral/cognitive changes, sphincter incontinence, and sexual dysfunction.40 Optic neuritis, a manifestation present in patient 4, as well as the sensitive symptoms and spinal cord involvement are rare manifestations, seldom mentioned in the literature.4 The parenchymal involvement corresponds to arterial occlusion or aneurysms and dural sinus thrombosis that leads to IIH.42–45 The first patient presented a confirmed case of IIH; whilst dural sinus thrombosis was not objectively documented, the clinical and CSF characteristics would be consistent with non-parenchymatous CNS involvement secondary to BD.
Vascular involvement. BD may affect both the arterial and venous system. The most important extravascular prototype lesions are: arterial occlusion, aneurysms, venous occlusion and thrombosis. The frequency is higher in young males and venous system involvement (80%) is more frequent than arterial involvement (20%).46 Deep venous thrombosis of the lower extremities is the most frequent vascular manifestation. Pulmonary artery aneurysms are the complication with the highest mortality described in Bd.4
GI Tract Involvement (GIT). It is characterized by inflammation of the mucosa leading to the development of single or multiple ulcers throughout the GI tract, with higher frequency in the ileocecal region.13 Clinically, it may present with abdominal pain, dysentery or hematochezia or acute abdominal pain due to visceral perforation. The differential diagnosis should be against Crohn’s disease, but it may impossible to differentiate in the absence of extraintestinal manifestations.6
As previously mentioned, there is not one pathognomonic sign or symptom in BD. The local epidemiology in terms of clinical manifestations is consistent with the findings in other populations. Due to the variability of the clinical spectrum and the asynchronous presentation of the manifestations, the diagnosis of BD requires a meticulous clinical judgement, since there are no definitive diagnostic histopathological or laboratory tests available. The diagnostic criteria for BD are listed in Table 2.
International criteria for the diagnosis of Behçet's disease (ICBD) 2014.
| Mouth ulcers | 2 points |
| Minor, major or herpes like ulcers identified by the physician or the patient, with a minimum of 3 episodes during a 12-month period | |
| Genital ulcers | 2 points |
| Ulcers or aphthous scars in the genital area, identified by the physician or the patient | |
| Ocular manifestations | 2 points |
| Anterior and posterior uveitis or retinal vasculitis diagnosed by the ophthalmologist | |
| Cutaneous manifestations | 1 point |
| Erythema nodosum, folliculitis, papulopustular lesions or acne-like nodules, observed by the physician in glucocorticoid naïve post-adolescent patients | |
| Vascular manifestations | 1 point |
| Arterial or venous thrombosis, superficial phlebitis or aneurysms | |
| Neurological manifestations | 1 point |
| CNS involvement | |
| Peripheral nervous system involvement | |
| Positive pathergy test (optional) | 1 point |
| Skin sensitivity characterized by the development of a sterile pustule, 24–48h after a needle skin prick, observed by a physician |
Sensitivity 97%, specificity 97% and diagnostic accuracy 97%. Score ≥4 is diagnostic.
Source: International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338-47).
The pathergy test included in the diagnostic criteria for BD is a highly specific study (98-100%) but with poor sensitivity (10-68%)47,48 and its positivity varies based on the population studied; it is usually positive in endemic BD countries, but it is just 20-30% in Americans and Europeans.3 In our setting, a negative pathergy test does not rule out the diagnosis, although if it is positive, it is highly suggestive of the disease; therefore, the recommendation is to do the test in every patient with a clinical suspicion of the disease.
In the presence of a clinical presentation suggestive of BD, it is mandatory to use diagnostic support tests such as the HLA typing study. For this condition, the most frequent association described is the HLA-B51 allele, present in up to 70% of patients with BD in endemic countries.49,50 HLA-B*51:01 is the subtype more strongly associated with BD. Other HLA class I and class II molecules have been less frequently involved in other populations, including: HLA-A*26, HLA-B*15, HLA-B*27:02, HLA-B*39:01, HLA-B*52, HLA-B*57, HLA-B*5, HLA-B*38, HLA-B*35, HLA-B*56, HLA-Cw1, HLA- HLA-Cw14, HLA-Cw15, HLA-Cw16, HLA-DRB1*04 and HLA-DRB1*07.7,51,52 Of the 4 cases discussed, 2 were positive for HLA-B*51 and 2 for HLA-B*35, confirming the genetic association described for this disease.
The treatment for BD is multidisciplinary, usually led by rheumatologist, and should be individualized based on age, type and severity of the affected organ or system. A T2T or Treat-to-Target strategy has not been defined and the key objectives are the remission of inflammatory exacerbation episodes and prevention of relapses.53 In general terms, the cornerstone of therapy for the acute episode is the use of GCS, either alone or in combination with other immunosuppressive agents.4 According to the recommendations of the EULAR 2018 guidelines for the treatment of BD,54 topical glucocorticoids are the first line therapy for oral and genital ulcers. Another option is thalidomide, but it has a high percentage of side effects. Colchicine is indicated for arthritis and for the prevention of mucocutaneous manifestations (particularly erythema nodosum and genital ulcers).55 When there is severe involvement of internal organs, high-dose GCS are indicated, with or without cyclophosphamide, followed by oral GCS for maintenance therapy and immunosuppressants (azathioprine, ciclosporin). Generally speaking, azathioprine is indicated for uveitis, deep venous thrombosis, GI tract and CNS involvement, and mucocutaneous manifestations resistant to other therapies. The use of biologics has been reserved for patients that fail to respond to the use of DMARDs and the clinical experience has been mostly with anti-TNFα.54 The effectiveness of adalimumab has been reported in case series, particularly for the treatment of ocular involvement (uveal) and less frequently for mucocutaneous manifestations (genital ulcers), GI tract, CNS (cerebral vasculitis) and bilateral pulmonary artery aneurysm.53 In 3 of the 4 cases. Adalimumab therapy was indicated due to failed therapeutic response to GCS, immunosuppressants or colchicine, particularly due to persistent articular and mucocutaneous symptoms resistant to conventional therapy. These cases responded favorable to Adalimumab.
ConclusionBD is a multisystemic EB vasculitis of unknown etiology and clinical evolution with exacerbations and remissions of unpredictable frequency and duration. Whilst the estimated prevalence in our environment is low, BD is a clinical entity with significant morbidity, and hence is a disease of special interest to rheumatology. The presence of oral and genital ulcers should be suspicious for BD in the context of a patient with probable autoimmunity, particularly when articular, ocular, cutaneous and neurological symptoms coexist. The isolated brain stem atrophy and the aneurysms of the pulmonary artery are rare manifestations but highly suggestive of this condition. A genetic study for HLA typing and the pathergy test are recommended as part of the clinical approach. Further sociodemographic characterization studies are required, with a larger number of patients in Colombia, to learn additional details about the behavior of this disease in our setting, with to view to shorten the time to diagnosis. In any case, due to the heterogeneity of the clinical presentation, this disease requires a multidisciplinary approach.
Conflict of interestsThe authors have no conflict of interests to disclose.
The following are Supplementary data to this article:
Please cite this article as: Padilla-Ortiz D, Chamorro-Melo M, Santos AM, Arias-Correal J, Reyes-Martínez V, Rueda JC, et al. Enfermedad de Behçet: un reto diagnóstico en reumatología. Descripción de una serie de casos y revisión de la literatura. Rev Colomb Reumatol. 2020;27:308–316.
