



Sudden unexplained death refers to sudden deaths that remain without a conclusive cause of death after a complete autopsy has been performed. These cases are classified as deaths of arrhythmic origin, without any alterations in cardiac structure. In recent years, biomedical advances have allowed progressive interaction between genetic research and the forensic field to perform post-mortem genetic analyses, the so-called molecular autopsy. These studies reveal familial genetic alterations that cause heart diseases associated with arrhythmic events and sudden death. Recent post-mortem studies identify genetic alterations as the most probable cause of death in approximately 30% of cases. The results obtained after these analyses also allow for a translation into the clinical field to be made, focused on the early identification of relatives at risk of syncope, as well as the adoption of therapeutic measures for prevention and personalised treatment.
La muerte súbita inexplicada hace referencia a los fallecimientos repentinos que quedan sin causa concluyente del episodio letal tras realizarse una autopsia completa. Estos casos se catalogan como fallecimientos de origen arrítmico, sin alteración estructural cardiaca. En los últimos años, los avances biomédicos han permitido la progresiva interacción entre la investigación genética y el campo forense para realizar análisis genéticos post mortem, la llamada autopsia molecular. Estos estudios ponen de manifiesto alteraciones genéticas familiares causantes de patologías cardiacas asociadas a eventos arrítmicos y muerte súbita. Estudios recientes post mortem identifican alteraciones genéticas como causa más probable del fallecimiento en alrededor de un 30% de los casos. Los resultados obtenidos tras estos análisis permiten también realizar una traslación al ámbito clínico focalizada en la identificación precoz de familiares a riesgo de síncope, así como la adopción de medidas terapéuticas para la prevención y el tratamiento personalizado.
The term sudden death (SD) refers to a sudden and non-violent death which occurs during the first hour from the onset of symptoms or, if it occurs in the absence of witnesses, when the deceased individual has been seen in good physical condition during the 24h prior to the fatal episode.1 Cases of SD, due to their particular characteristic of being unexpected and sudden, or due to the circumstances that may occur simulating a violent or accidental death or death from poisoning, legally belong to the field of forensic medicine. The medico-legal autopsy of these cases aims to establish the cause of death. In 5–30% of cases, depending on age and the criteria, the autopsy does not identify the conclusive cause of death (blank autopsy).2 It has been established that the highest percentages of blank autopsies occur in autopsies performed on deceased individuals from young age groups, while the percentage of SDs without a cause drops considerably in the older population. In these cases, and if an extracardiac cause has been ruled out, the deaths are classified as of cardiac origin, which has been called sudden cardiac death (SCD) of arrhythmic origin in individuals with a structurally normal heart. The term sudden unexplained death (SUD) syndrome was proposed for those cases in which there are no pathological findings in the autopsy, or no pathological or electrocardiographic history before death.3
In most cases, the autopsy identifies the cause of death, with cardiac abnormalities being responsible for almost 85% of SDs. These heart diseases are the leading cause of death in western countries.4 An incidence of SCD of 40–100/100,000individuals/year in the general population, and of 0.46–3.7/100,000individuals/year in the young population is estimated. This gives an approximate estimation of 1100–9000 annual deaths in Europe and 800–6200 in the USA.5,6 In individuals aged over 50 years, coronary artery disease is the main cause of SCD, while cardiomyopathies and channelopathies are responsible for SCD in the younger population.7 Cardiomyopathies are characterised as being structural changes to the heart and they are visible during an autopsy, either in the macroscopic or microscopic study. This is due to the fact that they are conditions in which the tissue changes are progressive and to the fact that no major anatomical changes are visible in the earliest stages of the disease.8 Channelopathies are heart diseases which cause malignant arrhythmias associated with SCD, but with no changes to the heart structure. In these cases, the cause of death usually remains unresolved after the autopsy. When there are no indications at all as to the cause of death, it is called a blank autopsy. The death is therefore classified as SUD.9 In these cases, the most probable cause of death is suspected to be a cardiac arrhythmia, resulting in the term “sudden arrhythmic death syndrome” (SADS).
Cardiac arrhythmias are responsible for around one million syncopal episodes annually, some of them leading to the death of the affected individual.10 The fatal complications derived from arrhythmias only manifest when there is a perfect interaction between environmental and genetic factors.11 The pathophysiological mechanisms which induce arrhythmias have still not been well defined, although alterations in genes which encode for cardiac proteins have been identified as high-risk factors in the pathogenesis of familial arrhythmias. Epidemiological studies on SUD show that the fatal episode may occur at any age and in both genders equally, despite the fact that there is an established trend that unexpected death occurs either during sleep or during a sporting activity, especially due to coronary artery disease in male adults over 50.12,13 Among young adults, SUD is generally related to drug abuse which triggers the cardiac arrhythmia, but more than half of deaths at these ages, especially in children under the age of 15 years, arise from undetermined natural causes.14 The main inducer in these very young age groups tends to be sporting activity.15 A heart health check in young athletes is therefore recommended.16 In infants less than one year old (sudden infant death syndrome [SIDS]), SUD is the main cause of death and tends to occur while they are sleeping.17,18 A special group, and one which has been studied in more detail in recent years, is that of patients diagnosed with epilepsy who suffer SUD (sudden unexpected death in epilepsy [SUDEP]). No conclusive cause of death or of traumatic death secondary to an epileptic seizure is discovered in the autopsy.19 In recent epidemiological studies in this SUDEP group, it has been observed that SUD is the main cause of death in young epileptic patients.20 There may be various mechanisms involved in SUDEP which, in isolation or in conjunction, result in SUD.21 Therefore, cardiac arrhythmias have a genetic basis, meaning that the term “molecular autopsy” refers to the genetic testing of post-mortem samples from blank autopsies.22,23
Post-mortem genetic testing or molecular autopsy aims to assist in the diagnosis of cases which remain unexplained after the autopsy. This testing makes it possible not only to explain the sudden death of a family member, but also to prevent a new case of SCD in the family. Some studies focused on this concept have recently been published, reporting that up to 30–35% of blank autopsies in young individuals can be explained by genetic disorders, while in older subjects the percentage drops to 15–20%.24–27 Despite this evident clinical translation and recommendation of international clinical guidelines,28 genetic advances are still not being routinely applied to SUD cases.
GeneticsThe last 20 years has seen major technological advances in the field of next generation sequencing (NGS), which make it possible to analyse a large number of genes, including exomes and genomes, more quickly and cost-effectively than the traditional Sanger sequencing method.29 The main challenge today is not the sequencing technology, but the genetic interpretation and clinical translation of the large quantity of data which are obtained after the NGS analysis. Most of the identified variants are classified as of uncertain/ambiguous significance. In this regard, the discussion and consensus of a group of experts (clinicians, geneticists, genetic counsellors) are fundamental to reach a correct interpretation which allows appropriate clinical measures to be adopted for each individual.
The studies conducted during the last five years using this NGS technology in post-mortem cohorts of SUD and/or SCD have identified new genes and genetic alterations which cause these arrhythmic disorders. Genetics is therefore a key tool for identifying the cause of SD.30 This cause tends to be a genetic alteration, which induces a change in the proteins responsible for generating the electrical activity of the heart in the ion channels of cardiac myocytes (channelopathies). This group of syndromes primarily comprises long QT syndrome (LQTS), Brugada syndrome (BrS), short QT syndrome (SQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT).31
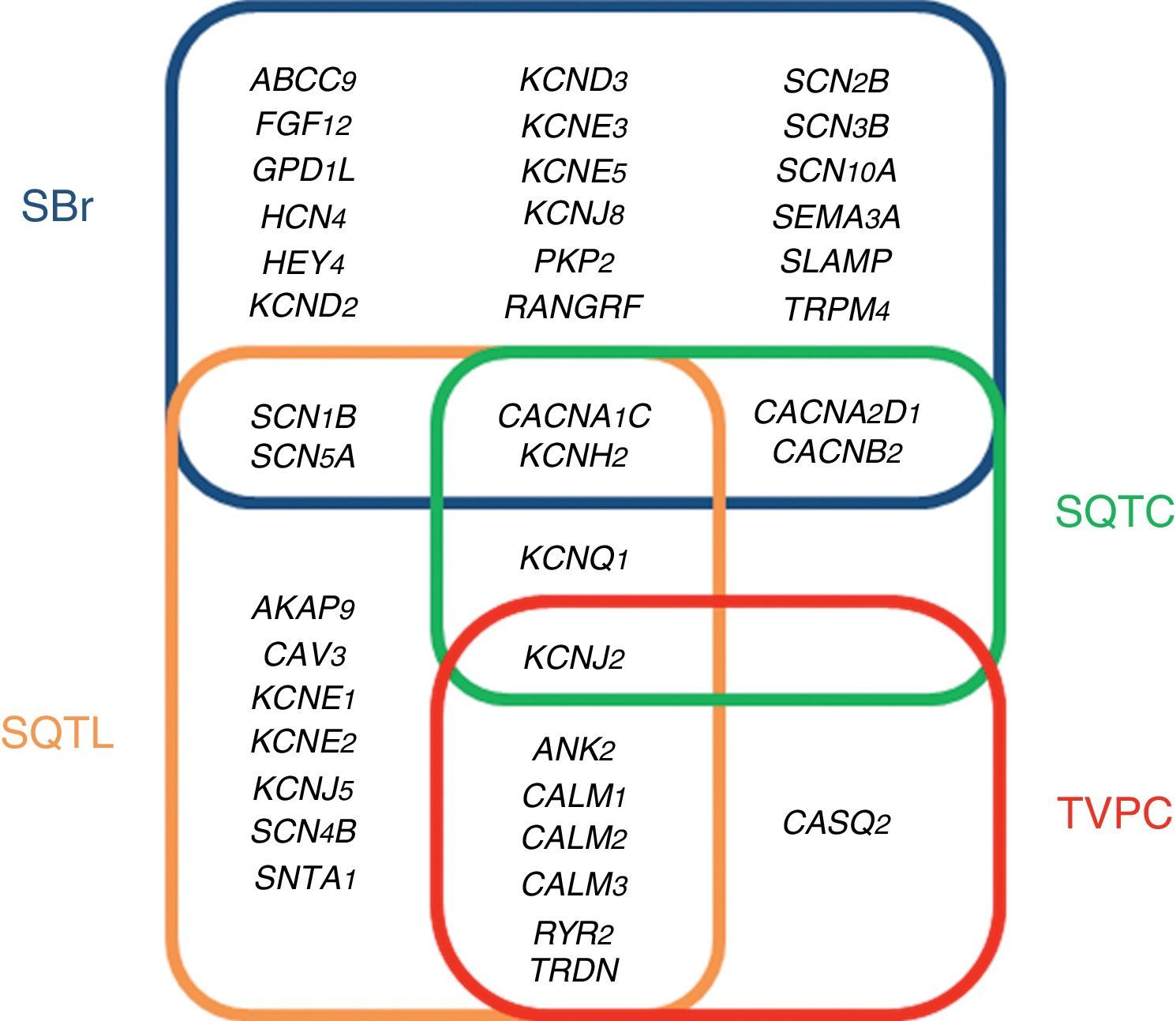
ChannelopathiesThe term cardiac channelopathy refers to a disease caused by alterations in some of the genes which encode for ion channels or their associated proteins (Fig. 1). These altered genes encode mainly for sodium (Na+), potassium (K+) and calcium (Ca2+) channels.32 In recent years, alterations in genes which encode for structural proteins of the desmosome of the cardiac myocyte, which may also induce this type of malignant cardiac arrhythmia, have been identified.33 The diagnosis is based on well-established electrocardiographic patterns, but the incomplete penetrance and the variable expressivity may hinder this diagnosis. The main risk of these arrhythmic syndromes is that the first clinical manifestation may be SCD itself. Early diagnosis is therefore essential for the identification of families with individuals at risk. As mentioned above, there are four main cardiac channelopathies that cause malignant arrhythmias and which may induce syncope.
Long QT syndrome
LQTS is electrocardiographically characterised by prolongation of the QT interval (QTc >480ms).34 The clinical presentation may range from asymptomatic patients to patients with polymorphic ventricular arrhythmias (torsades de pointes), the main cause of ventricular fibrillation and syncope. LQTS may be congenital or acquired. The latter is generally associated with drugs and water-electrolyte imbalance (www.torsades.org). The inheritance pattern may be either autosomal dominant (Romano-Ward) or recessive (Jervell and Large-Nielsen).35 Currently, more than 1000 genetic alterations located in 19 genes have been reported (AKAP9, ANK2, CACNA1C, CALM1, CALM2, CALM3, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ5, KCNQ1, RYR2, SCN1B, SCN4B, SCN5A, SNTA1 and TRDN).36 Genetic abnormalities causing QT interval prolongation characteristic of LQTS, either lengthen the duration of the action potentials by reducing the potassium current (loss of function) or by increasing the sodium or calcium current (gain of function). Despite the high number of mutations and genes identified as causing LQTS, 15% of families diagnosed with the disease still remain without a genetic cause after a thorough genetic analysis of all the known genes. Of the 85% of families with a genetic diagnosis, 35% show some alteration in the KCNQ1 gene (encoding for the protein KvLQT, which constitutes the alpha subunit of the slow rectifying channel [IKs]), 30% in the KCNH2 gene (encoding for the alpha subunit of the hERG channel which regulates the rapid potassium rectifying current [IKr]) and 10% in the SCN5A gene (encoding for the α subunit of the voltage-gated cardiac sodium channel Nav1.5, which regulates the late sodium current [INaL]).37 This means that the remaining 15 genes are responsible for only 10%. For this reason, the current clinical guidelines recommend only performing genetic testing for the three main genes.28
Brugada syndromeBrS is characterised by an atypical electrocardiographic pattern which shows right bundle branch block with ST segment elevation in the V1–V3 leads of the electrocardiogram, in the absence of structural cardiac changes, electrolyte disturbances or ischaemia.38,39 These changes in the electrocardiogram are often dynamic, showing themselves intermittently. They may not therefore be evident in the baseline electrocardiogram. A drug test for antiarrhythmic agents can unmask this diagnosis pattern.40 Symptoms of polymorphic ventricular tachycardia or ventricular fibrillation manifest mostly while at rest. Monomorphic ventricular tachycardia is a rare clinical presentation in BrS and is more prevalent in children, among whom the most common trigger is fever.41 The pathophysiology of this condition is multifactorial due to interaction among various genetic, hormonal and environmental mechanisms.42 Up to 2017, almost 300 alterations located in 24 different genes (ABCC9, CACNA1C, CACNA2D1, CACNB2, FGF12, GPD1L, HCN4, HEY2, KCND2, KCND3, KCNE3, KCNE5, KCNH2, KCNJ8, PKP2, RANGRF, SCN10A, SCN1B, SNC2B, SCN3B, SCN5A, SEMA3A, SLMAP and TRPM4), which follow an autosomal dominant inheritance pattern, have been identified.43 These genes encode for sodium, potassium and calcium channels and associated proteins which help in the functioning of these channels; recently, alterations in genes such as PKP2, which encode for plakophilin, a key protein in the desmosome of myocytes, have been reported.44 A complete analysis of all genes identifies the cause of the disease in 35% of cases, with the gene SCN5A being responsible for 25% of these. Therefore, despite the high number of reported genes, 65% of families diagnosed with BrS remain without a genetic diagnosis.43 The current clinical guidelines recommend only performing genetic testing of the gene SCN5A.28
Short QT syndromeSQTS is characterised by presenting a short QT interval in the electrocardiogram (QTc <330ms), with tall peaked T waves, and a non-prolonged interval between the peak and the end of the T wave. This syndrome was reported for the first time in 2000 and, from the start, it was associated with atrial tachyarrhythmias and very severe malignant ventricular tachyarrhythmias and SD at a young age.45 SQTS follows an autosomal dominant inheritance pattern.46 Genetic alterations have been reported in six genes, three of which are associated with potassium channels (KCNH2, KCNQ1 and KCNJ2)–gain of function in potassium channels–and the other three associated with calcium channels (CACNA1C, CANB2B and CACNA2D1)–loss of function in calcium channels. These effects induce a shortened repolarisation phase during the action potential and a shortening of the electrocardiogram QT interval. The main gene responsible for SQTS is KCNH2, accounting for almost 20% of all cases. After complete genetic testing, a genetic diagnosis is achieved for around 40% of cases, with more than half of diagnosed familial cases with an undetermined genetic cause for the disease.47 The current clinical guidelines recommend performing genetic testing of all genes except CACNA2D1, as its link with SQTS has not been studied in depth.28
Catecholaminergic polymorphic ventricular tachycardiaCPVT is an inherited arrhythmogenic syndrome characterised by the presence of bidirectional and polymorphic ventricular tachycardia following an adrenergic stimulus, such as exercise, emotional stress or excitement.48 A curious phenomenon is that the baseline electrocardiogram of patients with CPVT tends to be normal. Therefore, the diagnosis is based on the presence of arrhythmias during an effort test or using a Holter monitor. The clinical presentation of CPVT includes palpitations, dizziness and even seizures, which may trigger ventricular fibrillation and syncope. CPVT is a syndrome with a mortality rate of 30% to 50% in young age groups.49 There are eight genes which are related to CPVT: ANK2, CALM1, CALM2, CALM3, CASQ2, KCNJ2, RyR2 and TRDN. The first two follow an autosomal dominant inheritance pattern and the last two follow an autosomal recessive inheritance pattern. These four genes are responsible for 65% of diagnosed cases. This means that 35% of families with CPVT do not have a genetic diagnosis following thorough testing.32 These genes are involved in the regulation of intracellular calcium and increase the release of calcium from the sarcoplasmic reticulum. This excess calcium is related to alterations in the membrane potential of the sarcolemma with the onset of late depolarisations, causing predisposition to fatal arrhythmias.50 The main gene causing the disease is RyR2, which encodes for a cardiac ryanodine receptor, located in the sarcoplasmic reticulum, and it is responsible for 55% of genetic diagnoses. The current clinical guidelines recommend performing genetic testing for RyR2 and CASQ2.28
Molecular autopsyDespite the lack of standardisation in autopsy protocols regarding SUD, some consensus guidelines on this issue have been published in the last five years, in which the molecular autopsy has been included as part of the post-mortem analytical procedure. Examples include the Health Rhythm Society and the European Heart Rhythm Association (HRS/EHRA)51 or the Canadian Cardiovascular Society/Canadian Heart Rhythm Society,52 where genetic testing in cases of SUD, as well the testing of family members is recommended whenever a variant causing SUD is identified. Despite these recent breakthroughs, these guidelines still only recommend the analysis of the main genes associated with arrhythmic diseases, mainly for cost-effectiveness reasons.
Until relatively recently, the majority of diagnostic tests were carried out using Sanger sequencing technology. It is a very reliable, but slow and expensive technique, especially if the analysis of a high number of genes is desired. Over the past 10 years, new technologies (NGS) have been developed, which make it possible to sequence many genes simultaneously (even a whole exome or genome), in a few hours and at a reduced cost.53 The main problem with these NGS technologies is the large quantity of information that they provide us with, as we do not have the necessary tools to manage all these data or to reliably clinically interpret them.
Another important aspect is the taking of samples and their storage. In order to perform a molecular autopsy, DNA may be extracted from any biological tissue that has been stored in favourable conditions, but blood obtained within 48h of death is recommended. Freezing the EDTA tube with 5ml of blood at −20°C is recommended for correct storage of DNA. These freezing conditions should also be maintained when sending the tubes, avoiding sudden temperature changes. Thawing before DNA extraction should be gradual. Therefore, the Association for European Cardiovascular Pathology considers the correct storage of tissues and/or fluids in SUD autopsies to be essential for possible future testing.54 The Trans-Tasman Response Against Sudden Death in the Young (TRAGADY), supported by the Royal College of Pathologists of Australasia and the National Heart Foundation of New Zealand, have proposed a guideline to standardise the practice of autopsies in SUD in young people, secondary testing and the collection of material for post-mortem genetic testing (http://www.rcpa.edu.au/Library/Publications/Joint-and-Third-Party-Guidelines).
ConclusionsCardiac arrhythmic diseases cause a proportion of sudden deaths classified as unexplained after the autopsy. These diseases are of genetic origin. Therefore, the post-mortem testing of the genes associated with these electrical diseases of the heart identifies variants causing the disease in around 30% of cases. The new NGS tools for massive parallel sequencing enable this analysis to be performed quickly and cost-effectively. This molecular autopsy not only identifies the cause of death of the individual who died suddenly, but also the family members who are carriers of the genetic alteration, and who are therefore at risk of presenting with the disease and/or syncope. By identifying these family members at risk at an early stage, we can adopt the appropriate individualised monitoring, treatment and prevention measures.
Conflicts of interestNone declared.
Please cite this article as: Campuzano O, Sanchez-Molero O, Fernandez A, Iglesias A, Brugada R. Muerte súbita cardiaca de origen arrítmico: valor del análisis genético post mortem. Rev Esp Med Legal. 2018;44:32–37.