


La atrofia muscular espinal (AME) es una enfermedad neuromuscular de herencia autosómica recesiva con expresividad variable, que se caracteriza por degeneración y pérdida de las neuronas del asta anterior de la médula espinal y tallo cerebral, lo que resulta en debilidad muscular simétrica progresiva. La AME es debida a mutaciones homocigotas en el gen SMN1. El objetivo del presente estudio fue analizar clínica y molecularmente a cinco pacientes con AME, provenientes de dos familias no relacionadas. El análisis molecular se llevó a cabo mediante PCR y secuenciación automatizada, utilizando DNA genómico. El análisis mostró en ambas familias una deleción homocigota del exón 7 del gen SMN1. El presente estudio hace énfasis en la importancia de la realización de los estudios moleculares, ya que nos permite establecer el diagnóstico correcto del paciente.
Spinal muscular atrophy (SMA) is a neuromuscular disease of autosomal recessive inheritance with variable expressivity characterized by degeneration and loss of neurons in the anterior horn of the spinal cord and brainstem, resulting in progressive symmetrical muscle weakness. SMA is caused by homozygous mutations in the SMN1 gene. The aim of this study was to review the clinical and molecular characteristics of five patients with SMA from two unrelated families. Molecular analysis was performed by PCR and automated sequencing using genomic DNA. The analysis showed a homozygous deletion of exon 7 of SMN1 gene in both families. This study emphasizes the importance of performing molecular studies; allow the correct diagnosis of the patient.
¿ Introducción
La atrofia muscular espinal (AME) es una enfermedad neuromuscular de herencia autosómica recesiva con expresividad variable, que se caracteriza por degeneración y pérdida de las neuronas del asta anterior de la médula espinal y tallo cerebral, lo que resulta en debilidad muscular simétrica progresiva. Tiene una prevalencia de 1/10 000 nacidos vivos con una frecuencia de portadores de 1/40-60.1,2
La AME se clasifica en: tipo I o enfermedad de Werdnig-Hoffman, que corresponde al 50-70% de los pacientes con AME. Se caracteriza por presentar debilidad muscular grave, de forma generalizada al nacimiento o durante los primeros seis meses de vida, manifestada con hipotonía, parálisis flácida simétrica sin llegar a controlar los movimientos cefálicos. La función respiratoria se encuentra afectada debido a la debilidad de los músculos intercostales, sin embargo conservan la actividad del diafragma ocasionando una respiración paradójica. La denervación bulbar resulta en fasciculaciones de la lengua y dificultad para la succión y deglución, lo que disminuye la protección de las vías respiratorias y aumenta el riesgo de neumonía por aspiración. La muerte de estos pacientes suele ocurrir en los primeros dos años de vida, debido a falla respiratoria.1,3
La AME tipo II es la forma intermedia de la enfermedad, se caracteriza por inicio entre los 7-18 meses. Estos pacientes logran el sostén cefálico y la habilidad de sentarse por sí solos. Algunos llegan a ponerse de pie, pero no caminan sin ayuda. Usualmente, se desarrolla cifoescoliosis que requiere tratamiento ortopédico o quirúrgico. El tremor fino con la extensión de los dígitos o las manos empuñadas son comunes. La disfunción bulbar y la debilidad de los músculos intercostales ocasionan alteraciones en la deglución, con pobre ganancia de peso y dificultad para manejar las secreciones traqueales, causando esta última la mayoría de las muertes durante la adolescencia.1,3
La AME tipo III, también llamada enfermedad de Kugelberg-Welander es una forma leve con gran heterogeneidad clínica. Inicia durante la infancia o juventud, por lo que desarrollan la mayoría de las habilidades motoras. Algunos requieren asistencia para caminar o silla de ruedas durante la infancia, mientras que otros pueden tener sólo debilidad muscular leve y llevar una vida productiva adecuada. Frecuentemente desarrollan escoliosis y dolor articular, debido a la disminución de la fuerza muscular. La tipo IIIa inicia antes de los tres años de vida, y la IIIb a los tres años o más de edad. La AME tipo IV o tipo adulto es la más leve e inicia con sintomatología entre los 20-30 años de vida, con afección muscular sin problemas respiratorios o nutricionales.1,3
En algunos casos también se ha reportado la existencia de un tipo 0 o forma embrionaria, la cual se caracteriza por hipomotilidad fetal entre la semana 30-36 de embarazo, una debilidad muscular marcada asociada a artrogriposis.4
La AME es debida a la mutación del gen SMN1 localizado en 5q12.2-q13.3. Existe un segundo gen llamado SMN2, que difiere del SMN1 sólo por cinco nucleótidos. El gen SMN1 codifica una proteína denominada SMN (proteína de la supervivencia de la neurona motora), altamente conservada de expresión ubicua, se encuentra tanto en citoplasma como en núcleo y en este último se localiza en pequeñas estructuras llamadas gemas. La proteína SMN no se encuentra de forma aislada, sino en un gran complejo de proteínas ribonucleares, involucradas en diferentes aspectos del metabolismo del RNA. La SMN es esencial en la biogénesis de los snRNPs, llamados también riboproteínas pequeñas nucleares (U1, U2, U4, U4atac, U5, U11 y U12), las cuales son componentes críticos para el empalme del RNA, ya que ayudan a procesar el preRNA de SMN1, su alteración puede resultar en un empalme aberrante del preRNA.5
La proteína SMN mantiene la sobrevida de las neuronas motoras, lo que permite el transporte axonal normal y el mantenimiento de la integridad de la unión neuromuscular,6 su alteración puede causar degeneración de las neuronas motoras del asta anterior de la medula espinal, las cuales son especialmente vulnerables a las alteraciones de esta proteína.7
Los pacientes con AME tienen una pérdida homocigota del gen normal SMN1 generada por deleción, mutación puntual u otras, y mantienen al menos una copia del gen SMN2 (no se ha observado la pérdida de ambos genes, probablemente porque esto sería letal). En condiciones normales, el gen SMN2 sufre un empalme alternativo y produce una isoforma de mRNA, en el cual el exón 7 se encuentra ausente. La transición de C>T en el exón 7 (c.840C>T) es la responsable de este empalme alternativo, dando como resultado la isoforma SMNΔ7 no funcional, la cual es degradada rápidamente.
La ausencia del exón 7 del gen SMN1 se detecta en 95% a 98% de los afectados con AME, mientras que en el restante 2% a 5% se encuentran mutaciones a lo largo del gen SMN1, ya sea sin sentido, en sentido equivocado o mutaciones que afectan el marco de lectura. Además, se han reportado casos de heterocigotos compuestos. Aproximadamente, el 2% de los casos de AME son resultado de mutaciones de novo, de las cuales la mayoría ocurren en meiosis paterna.2 Estudios muestran que el número de copias de SMN2 puede modificar la severidad de la enfermedad, ya que aproximadamente 10% a 50% del pre-mRNA de SMN2 sufre un procesamiento adecuado y es subsecuentemente traducido a una proteína SMN de longitud completa, modificando así el fenotipo del paciente.1
En el presente estudio se analizan dos familias con diagnóstico de AME espinal tipos II y III, y se discuten los hallazgos clínicos y moleculares con lo reportado en la literatura médica.
¿ Material y métodos
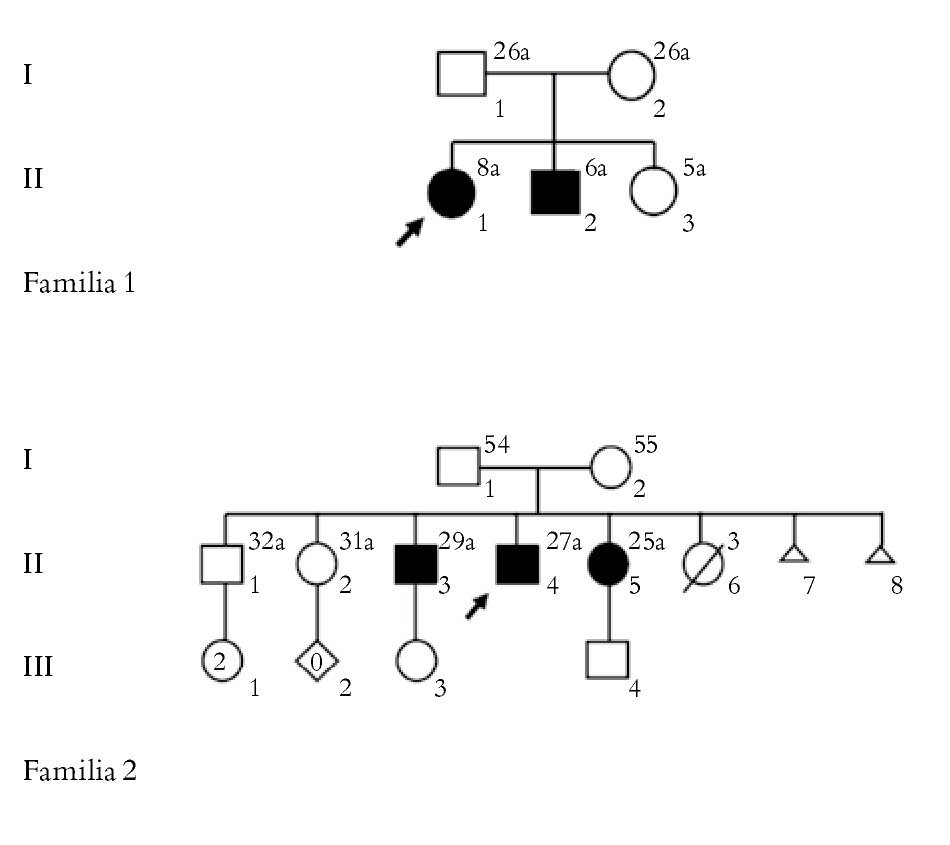
Se estudiaron tres pacientes clínica y molecularmente de cinco pacientes con manifestaciones clínicas compatibles con AME, provenientes de dos familias no relacionadas. Todos los pacientes fueron valorados por los Servicios de Neurología y Genética Médica del Hospital General de México "Dr. Eduardo Liceaga". En la Figura 1 se muestran las genealogías de las familias.
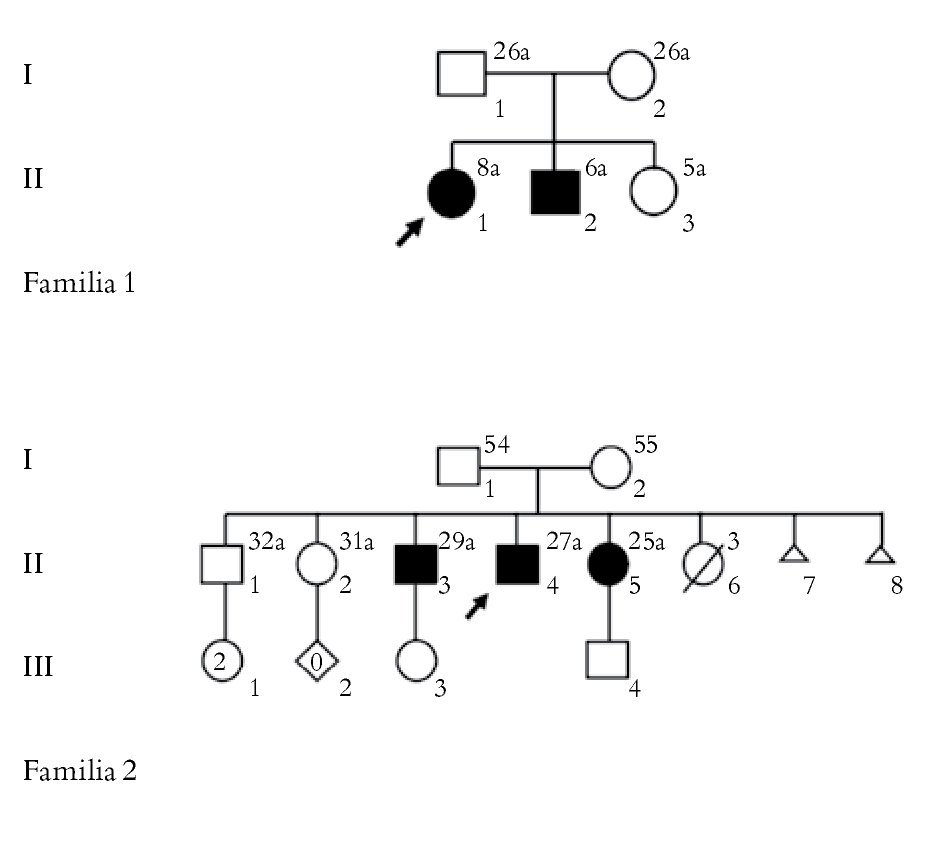
Figura 1. Árbol genealógico de las familias implicadas en el estudio.
Familia 1
La paciente II.1, del sexo femenino de ocho años de vida, es enviada por debilidad muscular progresiva de predominio en miembros pélvicos, producto de la primera gesta de padres sanos, sin antecedentes de importancia. Cursó con embarazo normoevolutivo, obtenida por cesárea por falta de progresión del trabajo de parto y APGAR 8/9. Presentó en el desarrollo psicomotor sostén cefálico a los tres meses, se sentó a los siete meses, posición de pie a los 11 meses, tiempo en el cual inicia su padecimiento con disminución de la fuerza muscular de predominio en miembros pélvicos. Actualmente, a la exploración presentó hipotrofia generalizada, extremidades superiores con disminución de la fuerza muscular 3/5, extremidades inferiores con hipotrofia, disminución de la fuerza muscular, ausencia de reflejos tendinosos profundos y sensibilidad conservada. No se observaron fasciculaciones linguales. Los estudios de laboratorio y la tomografía axial computada fueron normales. La electromiografía reportó inestabilidad de membrana y unidades motoras con amplitudes variables de 200uV a 8mV, con frecuencia de disparo incrementado y reclutamiento incompleto, con velocidades de neuroconducción normales. No se registraron fasciculaciones por lo que se consideró como probable AME tipo II.
El paciente II.2, del sexo masculino de seis años de vida, se presentó por disminución de la fuerza muscular de predominio en miembros pélvicos. Producto de la segunda gesta de padres jóvenes, aparentemente sanos, sin antecedentes de consanguinidad o endogamia. Cursó con embarazo normoevolutivo, obtenido por parto eutócico, APGAR 8/9. Su desarrollo psicomotor con sostén cefálico a los tres meses, se sentó a los siete meses. A esta edad inició con debilidad muscular progresiva, de predominio en miembros pélvicos, lo que ocasionó dificultad para mantener la posición de pie. A la exploración se encontró con hipotrofia generalizada, incapacidad a la marcha, fasciculaciones linguales, tórax asimétrico, disminución de la fuerza muscular en las cuatro extremidades, sensibilidad conservada. La electromiografía mostró ondas positivas, fibrilaciones en todos los músculos examinados y fasciculaciones. Se realizó neuro-conducción de nervio mediano cubital y tibial encontrando latencias sensoriales de amplitudes normales, latencias motoras normales, amplitud de potenciales disminuidos y velocidad de neuroconducción disminuida. El estudio con electrodo de aguja mostró datos de inestabilidad de membrana en todos los músculos examinados, con potenciales tipo neuropático. Todo estos datos fueron compatibles con AME tipo II.
Familia 2
La paciente II.4, del sexo masculino de 27 años de edad, es enviada por disminución de la fuerza muscular. No presentó antecedentes de consanguinidad o endogamia. Inició su padecimiento actual al año y medio de vida con disminución de la fuerza muscular en miembros inferiores, manifestada por caídas frecuentes que fueron aumentando hasta depender de silla de ruedas a los 15 años de edad. A la exploración se encontró con hipotrofia generalizada, fasciculaciones en extremidades superiores e inferiores, ausencia de reflejos tendinosos profundos y sensibilidad conservada. Los estudios de laboratorio fueron normales. La electromiografía reportó ondas positivas ++ en vasto medial bilateral, fasciculaciones en vasto medial bilateral, tibial anterior derecho, deltoides izquierdo y abductor corto del pulgar derecho, ondas con amplitud y duración aumentadas, velocidades de neuroconducción sensoriales normales, neuroconducción motora con latencias normales, disminución importante de la amplitud y velocidad de neuroconducción normal. Al estudio con electrodo de aguja se encontraron datos de inestabilidad de membrana, disminución de la inserción y presencia de fasciculaciones con patrón neuropático, siendo estos datos compatibles con AME tipo IIIa.
Análisis de DNA
Se extrajo DNA de sangre periférica por medio del método salino. Se evaluó la integridad del DNA por medio de electroforesis en gel de agarosa. Mediante la reacción en cadena de la polimerasa (PCR), se amplificaron los exones 7 y 8 del gen SMN1 con los siguientes oligonucleótidos, considerando la base de datos de NBCI del gen SMN1: F1 TTT CCT TAC AGG GTT TCA GAC; R1 CTT TCA TAA TGC TGG CAG AC, bajo las siguientes condiciones: se realizó la mezcla de reacción para un volumen final de producto de 25 µl a partir de una concentración de oligonucleótidos 1 mM, 20 ng de DNA, 2 mM de dNTPs, 1.5 mM de MgCl, 1 U de Taq polimerasa y buffer, según el proveedor con los siguientes parámetros: predesnaturalización 94°C/5 minutos, desnaturalización 94°C/1 minuto, alineamiento 50°C/2 minutos, extensión 72°C/3 minutos, elongación final 72°C/10 minutos, obteniéndose un amplicón de 95 pb. Se procesaron 3 µl del producto de PCR y se visualizaron en gel de agarosa teñido con bromuro de etidio, para la correcta visualización de los amplicones correspondientes a lo esperado (95 pb). La reacción de secuenciación se realizó siguiendo las indicaciones del proveedor (Applied Biosystem) en un secuenciador automatizado ABI 3100. Los polimorfismos de longitud de los fragmentos de restricción (RFLPs) se llevaron a cabo con la enzima DRA1 (que corta solamente en el exón 7 de SMN2), siguiendo las indicaciones del proveedor.
¿ Resultados
Familia 1
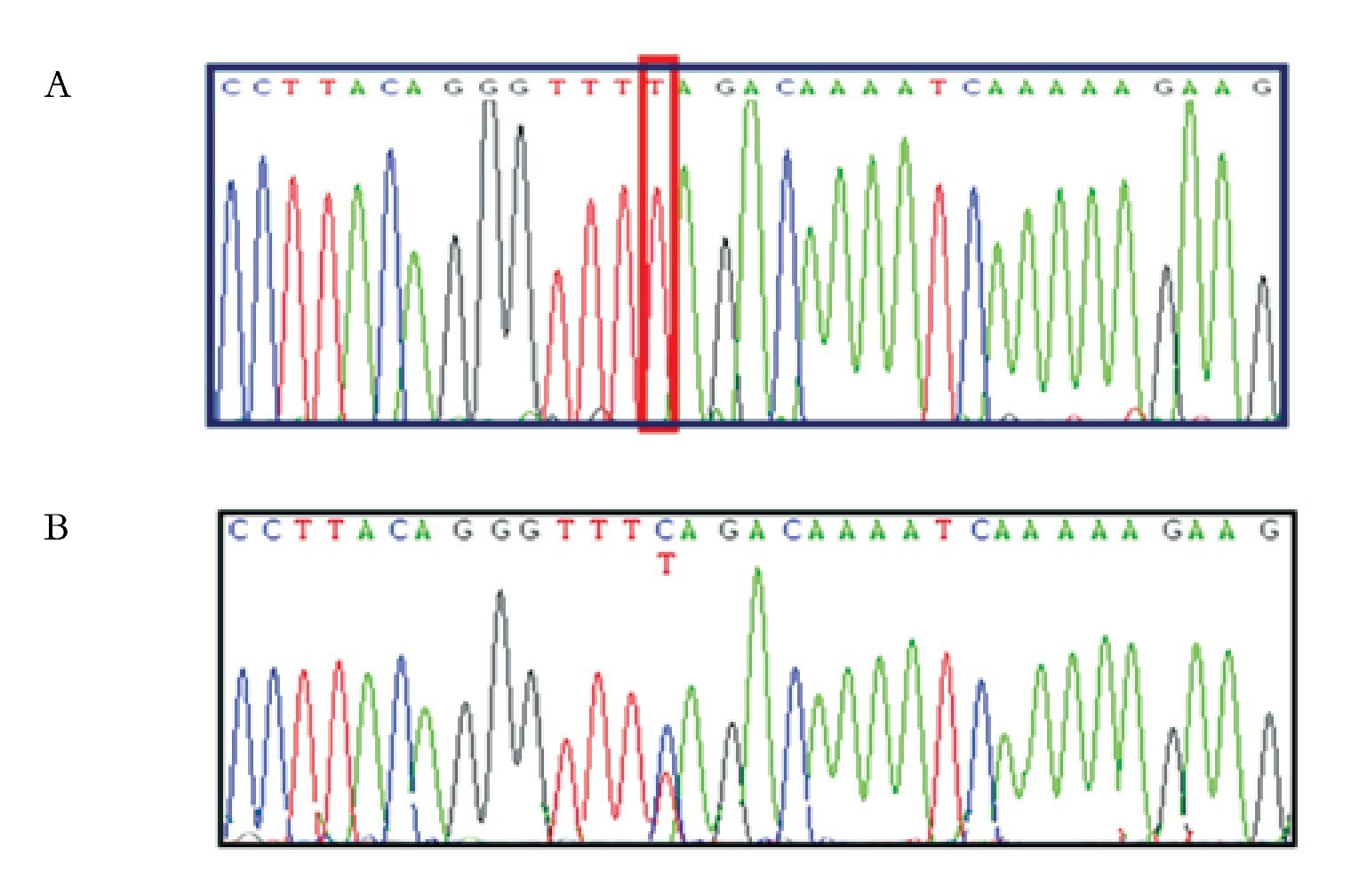
El estudio molecular en la paciente II.1 de los exones 7 y 8 de los genes SMN1 y SMN2, reveló únicamente la presencia de timina en el nucleótido 840 de los genes SMN1 y SMN2, indicando la pérdida en estado homocigoto del exón 7 del gen SMN1 (Figura 2), el cual tiene a la citosina en lugar de timina. El estudio molecular de la madre de la paciente II.2, mostró la citosina y timina en el nucleótido 840 del exón 7 de los genes SMN, indicando la presencia de ambos genes SMN1 y SMN2 (Figura 2).
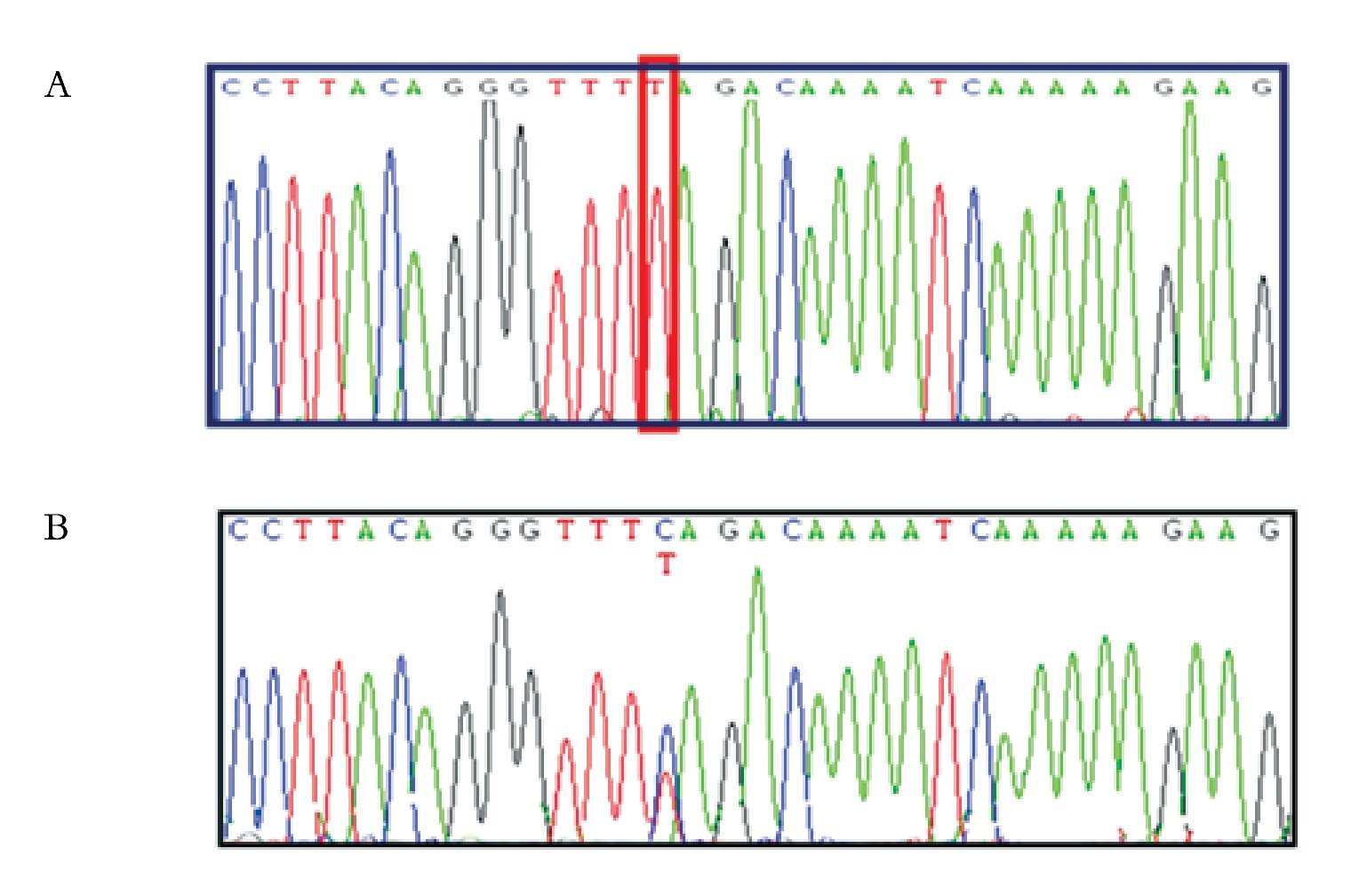
Figura 2. Electroferograma que muestra la secuencia parcial del exón 7 de los genes SMN1 y SMN2. A) Se observa la presencia de timina y ausencia de citosina en la posición 840 del exón 7 de los genes SMN (homocigoto), B) visualizando la presencia de citosina y timina en la posición 840 del exón 7 de los genes SMN (heterocigoto).
Familia 2
En la familia 2, se estudiaron molecularmente tres individuos (II.3, II.4 y II.5). El estudio molecular de los exones 7 y 8 de los genes SMN1 y SMN2 revelaron la presencia de timina únicamente (y por ende, la ausencia de citosina) en el nucleótido 840 del exón 7 de los genes SMN1 y SMN2, lo que es compatible con deleción homocigota del exón 7 del gen SMN1 (Figura 2).
Se realizó el estudio molecular de RFLPs con la enzima DRA1 en el paciente II.3 para confirmar los hallazgos, en busca de la presencia o ausencia de la deleción o exclusión del exón 7 del gen SMN1. Se confirmó que el método de secuenciación es útil para confirmar la pérdida del exón 7 en estado homocigoto de los genes SMN1 y SMN2, no así para identificar el número de copias del gen SMN2.
¿ Discusión
La falta de la proteína SMN ocasiona pérdida de las neuronas motoras del asta anterior de la médula espinal, lo cual desencadena debilidad muscular generalizada y progresiva de predominio en miembros inferiores, conservando la sensibilidad. En el presente estudio se analizaron dos familias con un total de cinco casos. Se realizó estudio molecular en todos ellos con sintomatología clínica de AME. Se confirmó la ausencia de citosina en la posición 840 de los genes SMN1 y SMN2, lo que se traduce en pérdida del exón 7 del gen SMN1 en estado homocigoto. Esto se explica porque la única variación entre ambos exones de los genes es la presencia de citosina en el gen SMN1, y de timina en el gen SMN2 en la posición 840. Por lo que consideramos que el diagnóstico molecular fue confirmatorio en este caso.
Los datos clínicos más consistentes en los pacientes fueron debilidad muscular progresiva, ausencia de reflejos tendinosos profundos, sensibilidad conservada y fasciculaciones. Los cambios electrofisiológicos fueron inestabilidad de membrana, reclutamiento incompleto o ausente en el estudio con electrodo de aguja monopolar, fibrilaciones, fasciculaciones, mientras que las velocidades de neuroconducción motora y sensitiva se encontraron sin alteraciones.
El estudio por medio de secuenciación detecta la deleción del gen SMN1 en estado homocigoto, sin embargo no proporciona información sobre el número de copias del gen SMN2 o sobre el estado de portador, aún así es posible diagnosticar 95% a 98% de los pacientes con AME, lo cual es importante tanto para el pronóstico como para el asesoramiento genético del paciente. Estudios previos muestran que el número de copias de SMN2 puede modificar la severidad de la enfermedad, ya que aproximadamente 10% a 50% del pre-mRNA de SMN2, sufre un empalme adecuado y es subsecuentemente traducido a una proteína SMN de longitud completa, modificando así el fenotipo del paciente.1 En la población el número de copias de SMN2 varía de cero a tres, y se ha reportado que 10% a 15% de la población sana no tiene ninguna copia de SMN2.1
Los pacientes con AME tipo II o III pueden mostrar más copias de SMN2 que la tipo I. El 95% de los pacientes con fenotipo de AME tipo I, suelen tener una o dos copias de SMN2, mientras que prácticamente todos los pacientes con AME tipo III tienen tres o más.8 Por otra parte, se ha observado la presencia de cinco copias de SMN2 y deleción homocigota de SMN1, en parientes no afectados de pacientes con AME, lo que demuestra que cinco copias de SMN2 son suficientes para compensar la ausencia de SMN1.2 Además del número de copias de SMN2, otros factores pueden modificar o influir en el fenotipo en cuanto a la gravedad de la enfermedad.9 Se han reportado deleciones del gen NAIP en pacientes con formas severas de AME y mutación homocigota de SMN1. Sin embargo, no se ha establecido aún un efecto directo de estas alteraciones sobre la enfermedad.10 La expresión de plastina 3 (PLS3) es importante para la axonogénesis y podría ser un modificador protector. Sin embargo, el rol exacto de la plastina 3 aún requiere ser confirmado.11,12
En este estudio, los casos con AME fueron confirmados por estudio molecular encontrándose que las características clínicas y electrofisiológicas coincidieron con los datos reportados en la literatura médica. Los pacientes que cumplían con los criterios clínicos y electrofisiológicos fueron homocigotos para la deleción del exón 7 del gen SMN1, lo que representa la mutación más frecuentemente observada en los pacientes con AME. No se caracterizó el número de copias del gen SMN2 en los pacientes con la deleción del exón 7 en estado homocigoto del gen SMN1, sin embargo se infiere que el número de copias del gen SMN2 pudo haber sido mayor en la familia 2, ya que presentan sintomatología de menor gravedad.
El abordaje multidisciplinario facilita el diagnóstico de estos pacientes, mientras que el estudio molecular nos da el diagnóstico definitivo, permitiendo así dar un asesoramiento genético adecuado a estas familias. El presente estudio hace énfasis en la importancia de la realización de los estudios moleculares, ya que nos permite establecer el diagnóstico correcto del paciente y por tal motivo un asesoramiento genético adecuado. Este tipo de pruebas de laboratorio son trascendentales, ya que la identificación correcta del padecimiento nos permite conocer la evolución y por tanto, establecer un manejo apropiado para los pacientes.
Correspondencia:
Dr. Sergio A. Cuevas Covarrubias.
Servicio de Genética, Hospital General de México "Dr. Eduardo Liceaga".
Dr. Balmis 148, Colonia Doctores, Delegación Cuauhtémoc.
C.P 06720. México D.F., México.
Correo electrónico: sercuevas@yahoo.com