




The IgG4-related disease (IgG4-RD) is a recurrent chronic fibroinflammatory disease, probably of autoimmune origin, recently recognised. Its diagnosis is based on a combination of clinical, radiological, serological, histopathological and immunohistochemical data. However, the histopathology is considered as the golden “standard” for diagnosis. In most of the cases, the sum of lymphoplasmacytic inflammatory infiltrate with abundant IgG4+ plasma cells, storiform fibrosis and obliterative phlebitis makes a reliable diagnosis. Patients usually show elevated serum IgG4 concentrations and respond well to steroid therapy. Nowadays, IgG4-RD has been described in almost every organ: pancreatobiliary tract, liver, salivary glands, nasopharynx, bone marrow, lacrimal gland, extra-ocular muscles and retrobulbar space, kidney, lung, lymph nodes, meninges, aorta, skin, breast, prostate, thyroid gland and pericardium.
La enfermedad relacionada con IgG-4 (ERIgG-4) es una enfermedad fibroinflamatoria crónica recurrente, probablemente de origen autoinmune, reconocida recientemente, cuyo diagnóstico se basa en una combinación de datos clínicos, radiológicos, serológicos, histopatológicos y de inmunohistoquímica. Sin embargo la histopatología se reconoce como el “standard” de oro para el diagnóstico; la suma de infiltrado inflamatorio linfoplasmocitario con abundantes células plasmáticas IgG4+, fibrosis esteriforme y flebitis obliterativa permite hacer un diagnóstico confiable en la mayoría de los casos. Los pacientes frecuentemente presentan concentraciones elevadas de IgG4 sérica y responden muy bien a la terapia esteroidea. En la actualidad se ha descrito ERIgG4 casi en todos los órganos: tracto pancreatobiliar, hígado, glándulas salivales, nasofaringe, médula ósea, glándula lacrimal, músculos extra-oculares y espacio retrobulbar, riñón, pulmón, ganglios linfáticos, meninges, aorta, piel, mama, próstata, tiroides y pericardio.
The IgG4-related disease (IgG4-RD) is a recurrent chronic fibroinflammatory disease, probably of autoimmune origin, recently recognised. Its diagnosis is based on a combination of clinical, radiological, serological, histopathological and immunohistochemical data. Nowadays, IgG4-RD has been described in almost every organ: pancreatobiliary tract, liver, salivary glands, nasopharynx, bone marrow, lacrimal gland, extra-ocular muscles and retrobulbar space, kidney, lung, lymph nodes, meninges, aorta, skin, breast, prostate, thyroid gland and pericardium.1
IgG4-RD was initially identified in the pancreas and was called autoimmune pancreatitis (AIP). Although it was made public in 1961, the first description of the AIP histopathological data was published in 19912,3 and in 2001 it was related to high levels of serum IgG4.4 The observation that patients with AIP have fibroinflammatory injuries rich in IgG4 plasma cells+extrapancreatic cells led to the IgG4-RD concept in 2003.5 During the following years, the spectrum of the disease broadened to include bile ducts, retroperitoneum and salivary glands. Besides, it was recognised that the extrapancreatic injuries can precede, coexist or appear after the AIP diagnosis. Nowadays, diseases such as Mikulicz's disease, Küttner's tumour, multifocal fibrosclerosis, inflammatory pseudotumour, Riedel's thyroiditis, aorta's inflammatory aneurysm, cutaneous pseudolymphoma and angiocentric fibrosis are considered part of the IgG4-RD spectrum.
The IgG4-RD nomenclature has evolved and has been known as IgG4-related systemic disease, IgG4-related sclerosing disease, IgG4-related autoimmune disease, hyper-IgG4 disease and IgG4 syndrome, among others. In 2011, an international committee, comprising 35 clinicians, pathologists and radiologists, gathered in Boston, Massachusetts primarily to establish norms for the histopathological criteria for the disease diagnosis.6 In 2012, a group of Japanese investigators suggested the name IgG4-RD, which has been accepted. The increase in serum IgG4 concentrations is useful in suspecting the IgG4-RD diagnosis, although they are neither enough nor necessary. Patients can have increases up to 25 times the normal IgG4 values in serum, or even higher; but between 20% and 40% have normal concentrations even when they show classical histopathological criteria of the disease.7 Serum IgG4 concentrations tend to be higher in patients with multiple organ involvement, and they are not indicators of disease activity or response to treatment. On the contrary, patients who are going through the last stages of the disease, when sclerosis is the prevailing histological datum, tend to show low IgG4 concentrations. The increase in serum IgG4 concentrations is not exclusively linked to IgG4-RD. There are other diseases such as atopic dermatitis, parasitary infections, pemphigus vulgaris, pemphigus foliaceus and pancreatic carcinoma which also show increases in this protein.8
IgG4 is the less common Ig subclass, encompassing approximately between 3% and 6% of the entire IgG fraction. It is unique due to its ability to join C1q, and therefore it cannot activate the classical pathway of complement activation.9
The IGg4-RD physiopathology is not clear yet. The disease can result in a hypersensitivity reaction/allergy or be of autoimmune origin. Some studies indicate that it is produced biphasically. The initial stage is induced as a response towards autoantigens such as carbonic anhydrase and lactoferrine as a result of the decrease of regulatory-natural T cells. Besides, the activation of cooperative T cells type 1 releases proinflammatory cytokines like interleukin 10, responsible for increasing serum IgG4 concentrations and stimulating plasma cells, which produce this immunoglobulin. The second stage is a result of a greater activation amount of memory regulatory T cells and type 2 cooperative T cells, which explains the chronic inflammation. The inflammatory infiltrate is formed by a mix of T and B cells, with an increase in T CD4+, CD 25+, and FOXP3+ cells. The cytokines produced by the type 2 cooperative lymphocytes (interleukine 4, interleukine 5 and interleukine 13) and regulatory cytokines (interleukine 10 and transforming growth factor) are overegulated in the involved tissues. Another hypothesis suggests an increase in the type 2 cooperative T cells response before the gut microflora or an immune response based on the presence of immune complex deposits in the basal membranes of the pancreatic acini and renal tubules.8,10
Patients with IgG4-RD are usually men over 50 years and tend to form nodular injuries in organs. These pseudotumours are usually misdiagnosed as malignant neoplasias, commonly affecting the orbit, salivary glands, lung, kidney, lymph nodes, retroperitoneum and other organs. The diffuse infiltrative disease was described in the meninges, skin, aorta or peripheral nerves. In some cases, only one or two organs are affected, and in other cases there is multiple organ involvement.11 Multiple organ injuries can appear synchronously or metachronously.12 In the series with higher number of cases (114), published so far by Zen et al., 31% of them showed systemic or multiple organ disease; 24% showed pancreatobiliary disease; in 20% the disease was located in head and neck; 14% showed thoracic disease and 11% retroperitoneal disease.10
Patients with IgG4-RD commonly have history of allergic rhinitis, sinusitis, asthma and other allergic or atopic diseases. Some patients show serum IgE increases and peripheral eosinophilia.
This disease has an indolent clinical course with manifestations which can take months or years to appear. The diagnosis takes around 13.5 months to be made (4–60 months).13 Most of the patients often show general symptoms and usually feel well despite having a systemic disease. However, a small group of subjects have an acute disease characterised by general symptoms, fever and serum increase in acute phase reactants.6
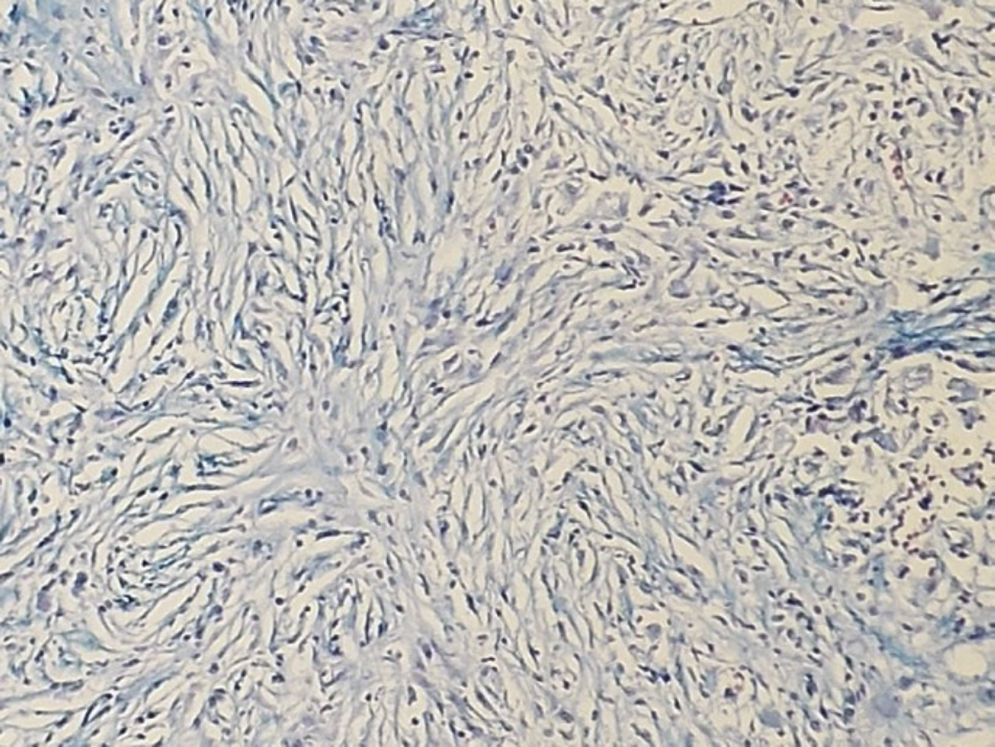
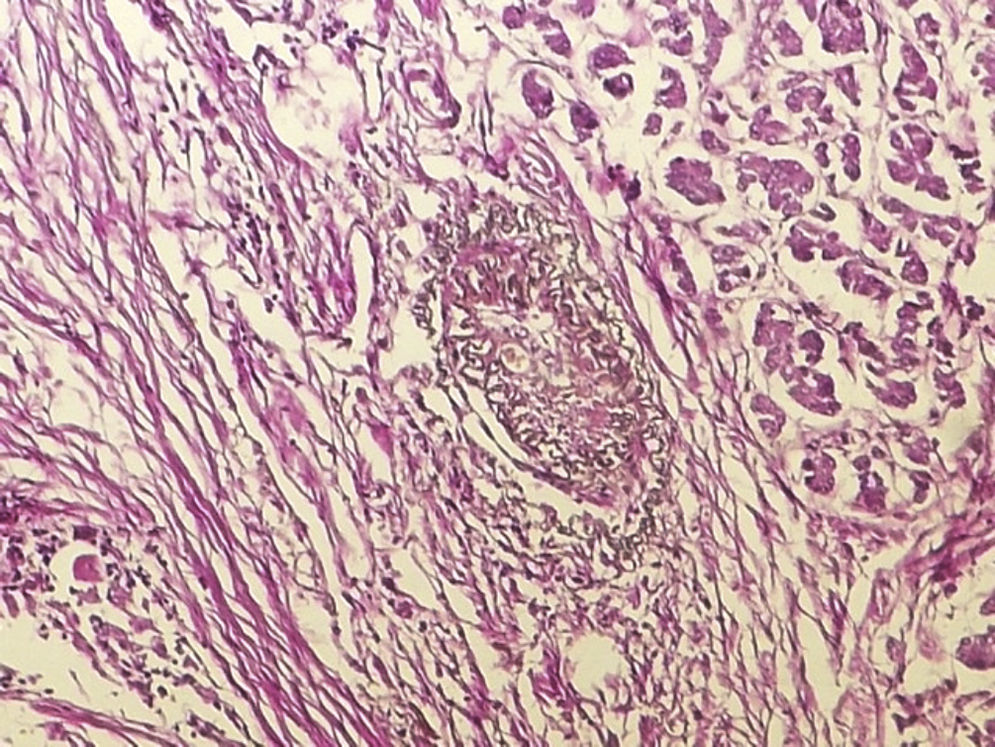
IgG4-RD diagnosis not only requires a characteristic histopathological appearance but also a high number of IgG4+ plasma cells. The three histopathological criteria are: lymphoplasmacytic inflammatory infiltrate, fibrosis arranged, at least focally, in storiform pattern (Fig. 1) and obliterative phlebitis (Fig. 2). These changes cause atrophy and loss of the specialised structures from the different tissues.14 The tissues show similar changes regardless of the involved organ.1,15


At the beginning, it was believed that the number of IgG4+ plasma cells was a highly specific marker of the disease; however, it is now known that this finding is unspecific. Diseases such as ANCA-associated vasculitis, rheumatoid arthritis, inflammatory bowel disease, Rosai-Dorfman disease, autoimmune atrophic gastritis, Castleman disease, primary sclerosing cholangitis, reactive perforating collagenosis, inflammatory myofibroblastic tumour and some carcinomas can present an increase in the number of positive IgG4 plasma cells.16,17 Today it is recommended that a quantitative analysis of the positive plasma cells in the different organs be made and compared with the cut points established by the experts in 2012.16,17 The cut points vary from over 10 positive plasma cells in pancreas, liver or kidney biopsies to over 200 in cutaneous biopsies. However, it is important to emphasise that the number of positive cells should not be used as an isolated diagnostic criterion, because there are cases in which a diagnosis with a low amount of positive IgG4 plasma cells is made, particularly when there is advanced sclerosis.
Some investigators have suggested that if the proportion between IgG4+ and IgG+ plasma cells is higher than 40%, the IgG4-RD diagnosis can be made.14 However, this datum was not considered sufficient IgG4-RD pathological evidence at the experts’ consensus whose conclusions were published by Deshpande in 2012.17 There are cases that show IgG4/IgG proportions >50% without the histopathological criteria, for instance, conditions in which there are high interleukine 6 serum concentrations, as in rheumatoid arthritis and Castleman disease.18
The combination of common histopathological data and a high number of IgG4+ plasma cells is the gold standard for diagnosis; however, in the appropriate context, there is enough evidence to make the IgG4-RD clinical-pathological diagnosis in the tissue examination that only shows some data.19 Specific terminology was suggested for the IgG4-RD diagnosis, including 3 categories: (a) Highly suggestive IgG4-RD histological data with at least 2 out of 3 histological data, IgG4+ plasma cell count according to the established parameters18 and IgG4+/IgG+ proportion >40%, (b) Histological data of probable IgG4-RD cases with only a histopathological datum (generally lymphocitary inflammatory infiltrate) and appropriate count of IgG4+ plasma cells. Clinical, radiological and serological evidence is required in these cases to confirm the diagnosis. This evidence can include: serum IgG4>135mg/dl and another organ involvement proved radiologically or pathologically, (c) Insufficient histological data for the IgG4-RD diagnosis: the biopsies in this category show lymphoplasmacytic inflammatory infiltrate without an increase in the number of plasma cells or fibrosis without storiform pattern16,17 (Table 1).
Terminology for the IgG4-RD histological diagnosis.
| Highly suggestive IgG4-RD histological data |
| 2 out of 3 histological data |
| IgG4+ plasma cells |
| IgG4+/IgG+ cells proportion>40% |
| Histological data of likely IgG4-RD |
| 1 histological datum |
| IgG4+ plasma cells |
| Serum IgG4>135mg/dl or other organ involvement, radiologically or anatomopathologically proven |
| Insufficient histological data for the IgG4-RD diagnosis |
| Lymphoplasmacytic inflammatory infiltrate |
| IgG4− plasma cells |
| Fibrosis without storiform pattern |
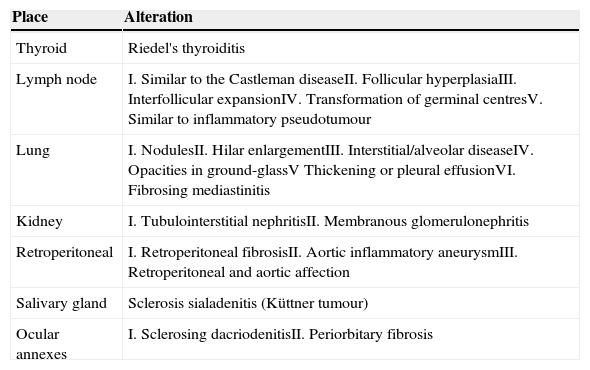
The involvement of the thyroid gland as part of the IgG4-RD spectrum is increasingly recognised because some of the patients present hypothyroidism and high levels of antithyroglobulin antibodies. Riedel's thyroiditis was also identified as an IgG4-RD manifestation.20 The thyroid biopsies of those patients in whom the disease is new show characteristic histopathological data and fibrosis when the IgG4-RD has evolved. Recently, a variant of Hashimoto's thyroiditis which is part of the IgG4-RD spectrum was described and can be the cause of the hypothyroidism appearing in some of these patients.21
The lymphadenopathy related to IgG4-RD can appear before, together with or after the diagnosis of the disease. Although several ganglionic groups are generally affected, predominantly the mediastinal, abdominal and axillary, patients do not usually show general symptoms; but they can have polyclonal hypergammaglobulinaemia, serum IgG4 concentrations and increase in serum IgE and IgG. It is common to take biopsies in these cases with presumptive diagnoses of lymphoma, Castleman disease or metastasic lymphomas. The histopathology is considerably variable in these cases; 5 patterns were described: (I) similar to Castleman disease, (II) follicular hyperplasia, (III) interfollicular expansion, (IV) progressive transformation of the germinal centres and (V) similar to inflammatory pseudotumour. A high number of IgG4 cells is observed in every pattern and the IgG4+/IgG proportion is >40%.8,22,23
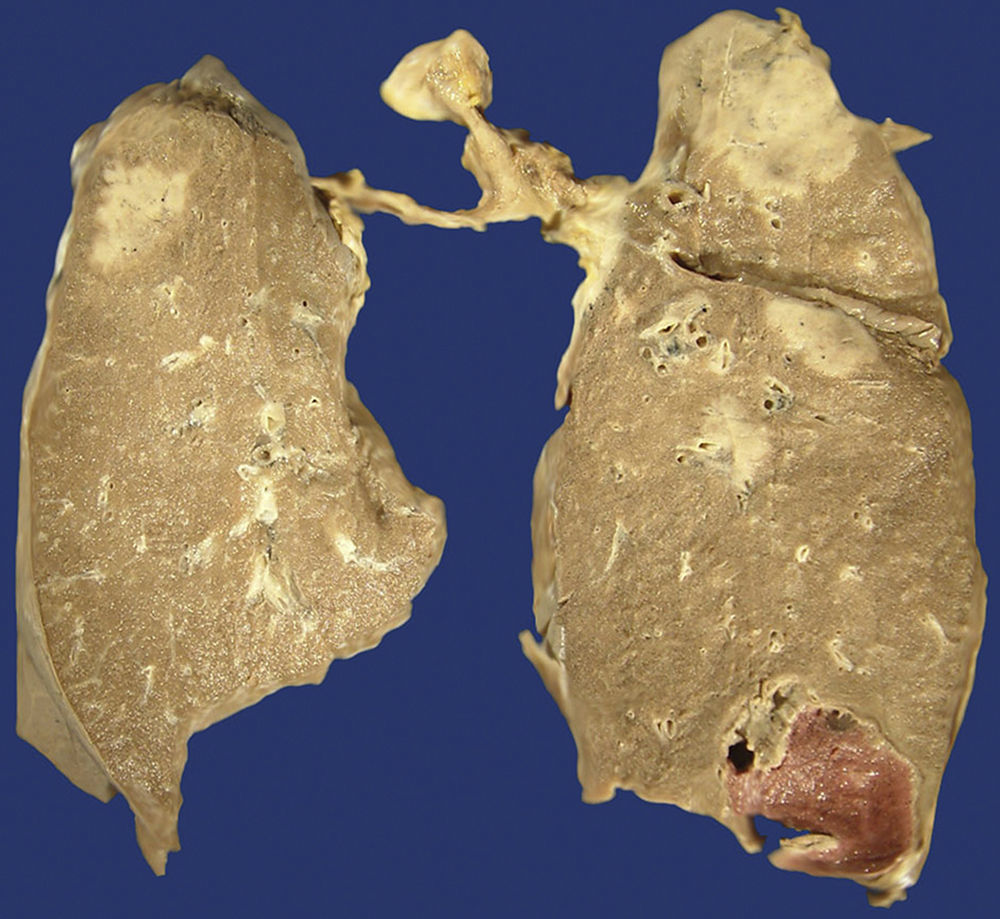
Approximately half of the patients with pulmonary disease related to IgG4 show cough, haemoptysis, dyspnoea, pleural effusion and chest pain. In other patients, the disease is asymptomatic and it is only discovered through image scanning. There are 6 morphological patterns described from the radiological and histopathological point of view: (1) nodules (Fig. 3), (2) hilar enlargement, (3) interstitial/alveolar disease with honeycomb pattern, diffuse images in ground-glass and bronchiectasis, (4) round opacities in ground-glass, (5) thickening or pleural effusion and (6) extrinsic compression of the upper airways by fibrosing mediastinitis.8,22,24

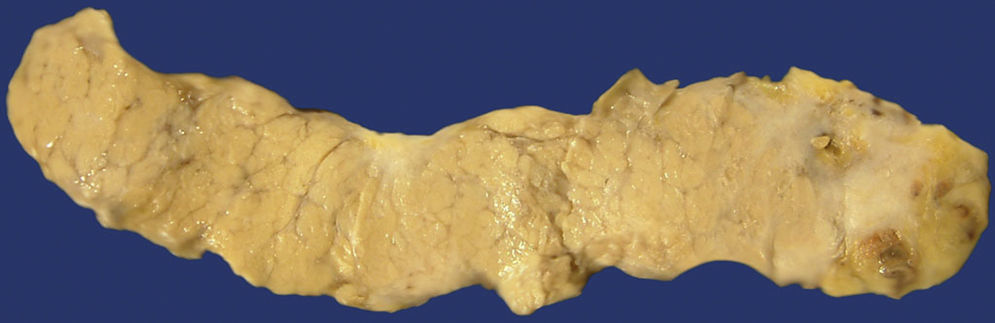
The symptomatology of patients with AIP differs from the symptomatology of those who suffer from other types of pancreatitis, particularly the alcoholic type. These patients usually have obstructive jaundice, loss of weight and mild or moderate abdominal discomfort. These clinical data, together with the radiological ones, such as occupative masses, frequently determine the suspicion of pancreatic carcinoma diagnosis (Fig. 4). Most of the cases are diagnosed by the characteristic tomographic findings described as “sausage-shaped pancreas” and are confirmed through biopsy with a thin needle or through endoscopic retrograde cholangiopancreatography.8,22,25

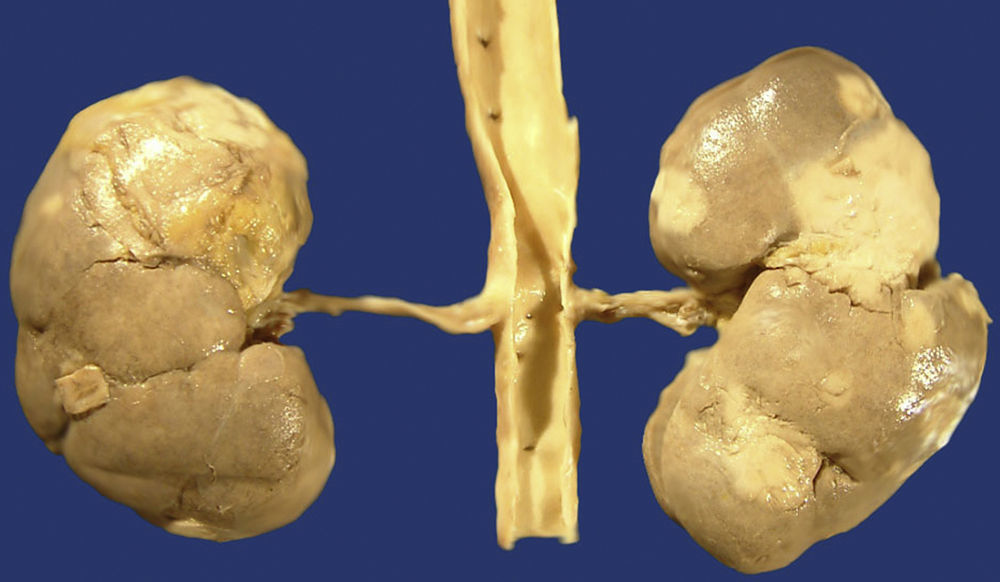
The IgG4-RD can also involve the kidneys and the urinary tract. In the kidneys, the most common symptom is the tubulointerstitial nephritis (TIN); only 7% of the patients with renal failure have membranous glomerulonephritis (MGN). Most of the patients with tubulointerstitial nephritis have acute or progressive renal failure. It appears as renal mass in some patients and both appear in other patients. Approximately 80% of the patients with TIN have an increase in the IgG or IgG4 serum levels. Other common laboratory findings are the decrease in the C3 and C4 levels, peripheral eosinophilia, urine protein, microscopic haematuria and low levels of antinuclear antibodies. From the radiological point of view, injuries are usually bilateral, multiple and predominant in the cortex, even though diffuse nephromegaly can also be observed (Fig. 5). In the biopsy or nephroctomy, lymphoplasmacytic inflammatory infiltrate rich in IgG4+ plasma cells, fibrosis and tubular atrophy is observed. The presence of IgG4 deposits was proven in the tubular basal membranes. Those patients with MGN associated to IgG4 show urine protein in nephritic ranges; in them, glomerulopathy is secondary to the deposit of immune complexes and not a consequence of the inflammatory process which characterises the IgG4-RD.8,22,26

The retroperitoneal fibrosis is one of the IgG4-RD manifestations. The abdominal aorta is usually involved in this condition and it can adopt two macroscopic patterns: one in the form of nodules or masses, and another in the form of plaques. Based on the sites affected by this form of disease, it can be classified as: (1) retroperitoneal fibrosis, (2) inflammatory aneurysm of the aorta, (3) a combination of retroperitoneal and aortic involvement and (4) chest aortitis.8,22,27
Most of the cholecystitis cases related to IgG4 are asymptomatic and were found incidentally. Approximately 25% of the AIP cases are associated to lymphoplasmacytic acalculous cholecystitis rich in IgG4+ cells with predominantly extramural involvement.8,22
The IgG4-RD of the salivary glands is shown as hard unilateral or bilateral tumoural masses which are frequently confused with neoplasic injuries. The patients are adults with an average age of 60 years, men being slightly predominant. In most of the cases, the submandibular gland is affected (Fig. 6). In the bilateral cases, the disease is generally asynchronous, even though it can be metachronous. Allergic rhinitis and asthma are common in these patients, and they usually have peripheral eosinophilia and high levels of serum IgG and antinuclear antibodies. Most of the patients show an increase in the serum IgG4; however, up to 40% can show normal levels of this immunoreactant.28
.")
Patients with ocular annexes affection show similar findings to the salivary gland affection. Its average age is 50 years; but there is no predominant genre. They appear with oedema or elevated painless unilateral or bilateral, periorbital or palpebral volume. A small number of them show diplopia or proptosis; however, visual acuity is rarely affected. The disease affects the lacrimal glands and the periorbital soft tissues. It rarely affects the conjunctiva28 (Table 2).
| Place | Alteration |
|---|---|
| Thyroid | Riedel's thyroiditis |
| Lymph node | I. Similar to the Castleman diseaseII. Follicular hyperplasiaIII. Interfollicular expansionIV. Transformation of germinal centresV. Similar to inflammatory pseudotumour |
| Lung | I. NodulesII. Hilar enlargementIII. Interstitial/alveolar diseaseIV. Opacities in ground-glassV Thickening or pleural effusionVI. Fibrosing mediastinitis |
| Kidney | I. Tubulointerstitial nephritisII. Membranous glomerulonephritis |
| Retroperitoneal | I. Retroperitoneal fibrosisII. Aortic inflammatory aneurysmIII. Retroperitoneal and aortic affection |
| Salivary gland | Sclerosis sialadenitis (Küttner tumour) |
| Ocular annexes | I. Sclerosing dacriodenitisII. Periorbitary fibrosis |
In the last years, great progress has been achieved regarding the IgG4-RD understanding; but much is still unknown about the clinical-pathological manifestations of the IgG4-RD. The diagnosis not only requires the presence of characteristic histopathological data but also the elevated IgG4. The correlation between the clinical, radiological and microscopic data is essential to arrive at the appropriate diagnosis.
Most of the studies of this disease come from Asia, and it is unknown whether there are clinical or pathological differences with the cases of western countries. There are some lymphoma cases associated with IgG4-RD; but more studies are required to confirm the possibility of a higher risk to suffer from lymphoma in patients with IgG4-RD.
Conflict of interestThe authors declare that they have no conflict of interests.