





El reumatismo fibroblástico (RF) es una enfermedad de aparición excepcional y de causa desconocida, caracterizada por la asociación de una poliartritis agresiva, esclerodactilia y la presencia de nódulos cutáneos. La artritis se caracteriza por ser rápidamente progresiva, dando lugar a la pérdida de la función articular con contractura flexora de los dedos. Las lesiones cutáneas se caracterizan por unos hallazgos histológicos característicos, por lo que su diagnóstico es fundamentalmente anatomopatológico. La biopsia de los nódulos y de la piel engrosada demuestra un incremento del espesor de las fibras colágenas y aumento de fibroblastos. Su diagnóstico diferencial incluye enfermedades como la artritis reumatoide, la esclerodermia nodular, la reticulohistiocitosis multicéntrica y otras tantas. Su tratamiento no está aún consolidado, aunque la mayoría de los casos han sido tratados con prednisona y metotrexato, con buenos resultados. Su pronóstico es generalmente bueno, sin repercusiones funcionales, sobre todo si el diagnóstico y el tratamiento se realizan de forma precoz.
Fibroblastic rheumatism is a rare disease of unknown cause and is characterized by the association of aggressive polyarthritis, sclerodactyly and the presence of cutaneous nodules. The arthritis is rapidly progressive, leading to loss of joint function with flexion contractures of the fingers. The cutaneous lesions are characterized by singular histological findings and consequently their diagnosis is mainly histopathological. Biopsy of a nodule or thickened skin typically reveals increased thickness of collagen fibers and fibroblastic proliferation. The differential diagnosis includes diseases such as rheumatoid arthritis, nodular scleroderma, and multicentric reticulohistiocytosis, among others. Treatment has not yet been consolidated, although most cases have been treated with prednisone and methotrexate with good results. The prognosis is generally good, without functional repercussions, especially if early diagnosis and treatment are provided.
El reumatismo fibroblástico (RF) se manifiesta principalmente por la asociación de poliartritis erosiva —de rápida progresión— y nódulos cutáneos no dolorosos de consistencia firme, con una histología característica (incremento del espesor de las fibras colágenas y aumento de fibroblastos)1. El diagnóstico es eminentemente clínico, aunque su confirmación viene de la mano del estudio anatomopatológico2. Se han utilizado varios fármacos para su tratamiento, y los más empleados han sido la prednisona y el metotrexato3–7. La irreversibilidad de los daños articulares ocasionados y su rápida progresión hacen necesario un diagnóstico precoz de esta entidad y optimizar el tratamiento utilizado.
Etiología y patogeniaEl RF es una enfermedad de aparición excepcional y de causa desconocida. Desde su descubrimiento en la literatura francesa por parte de Chaouat et al. en 19808, las referencias sobre esta enfermedad han sido muy escasas, de tal modo que su diagnóstico y tratamiento se basan en las experiencias de determinados autores, basada en casos aislados y no en una adecuada evidencia científica. Zelger y Burdorf9 propusieron que podría estar relacionada con la histiocitosis de células no-Langerhans, y otros autores la relacionaron con la reticulohistiocitosis multicéntrica10–15. Histológicamente se ha relacionado con el xantogranuloma o la histiocitosis nodular progresiva3,12,13.
Aunque, como hemos comentado, su patogenia es aún desconocida, en biopsias sinoviales se ha observado que la proliferación de fibroblastos y miofibroblastos juega un papel muy importante en el desarrollo de la fibrosis en piel y sinovial1,16,17. De este modo, se ha observado que los miofibroblastos están implicados en otras entidades de características fibróticas, tales como la fibrosis pulmonar y la fibrosis renal en la nefropatía IgA18. La presencia de macrófagos y linfocitos en la fase inflamatoria inicial sugiere que citoquinas fibrogénicas, tal como el factor de crecimiento tumoral beta (TGF-β), puedan estar implicadas en la proliferación fibroblástica y en la diferenciación miofibroblástica1,16,18 y promover la fagocitosis de colágeno por parte de los miofibroblastos18,19.
EpidemiologíaNo hay diferencias en cuanto a su distribución por sexo y el rango de edad es muy variable (8-68 años), con una media en torno a los 34 años. Es más frecuente en adultos, y los casos pediátricos son muy escasos20,21. De los 26 casos publicados en la literatura médica internacional, solo 5 casos han afectado a niños3,22–24. Su distribución es mundial, aunque la raza caucásica es la más frecuentemente afectada20; en solo un caso la procedencia era china7. No se han descrito casos asociados a antecedentes familiares, con lo que no existen datos contundentes para pensar en una posible herencia genética25.
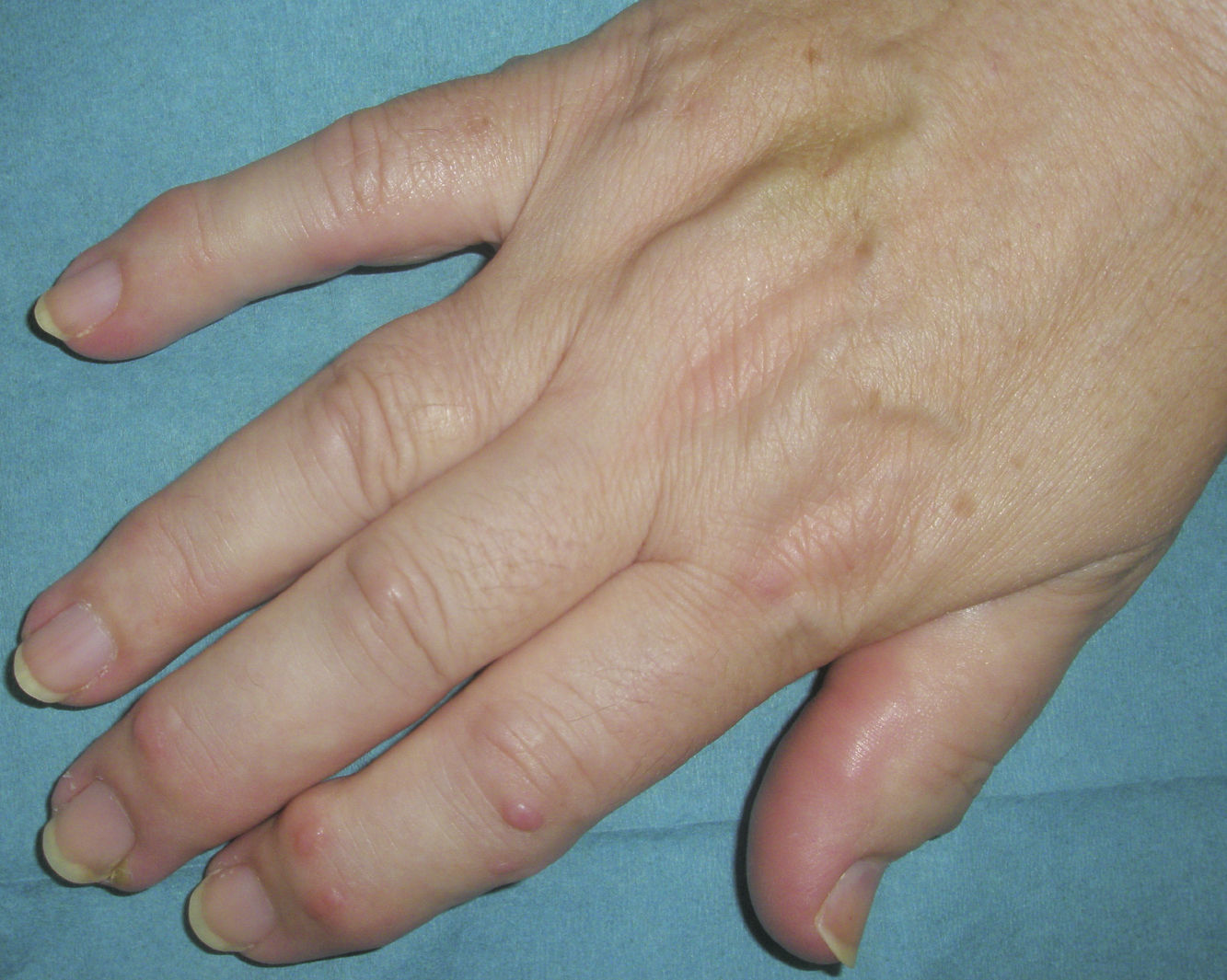
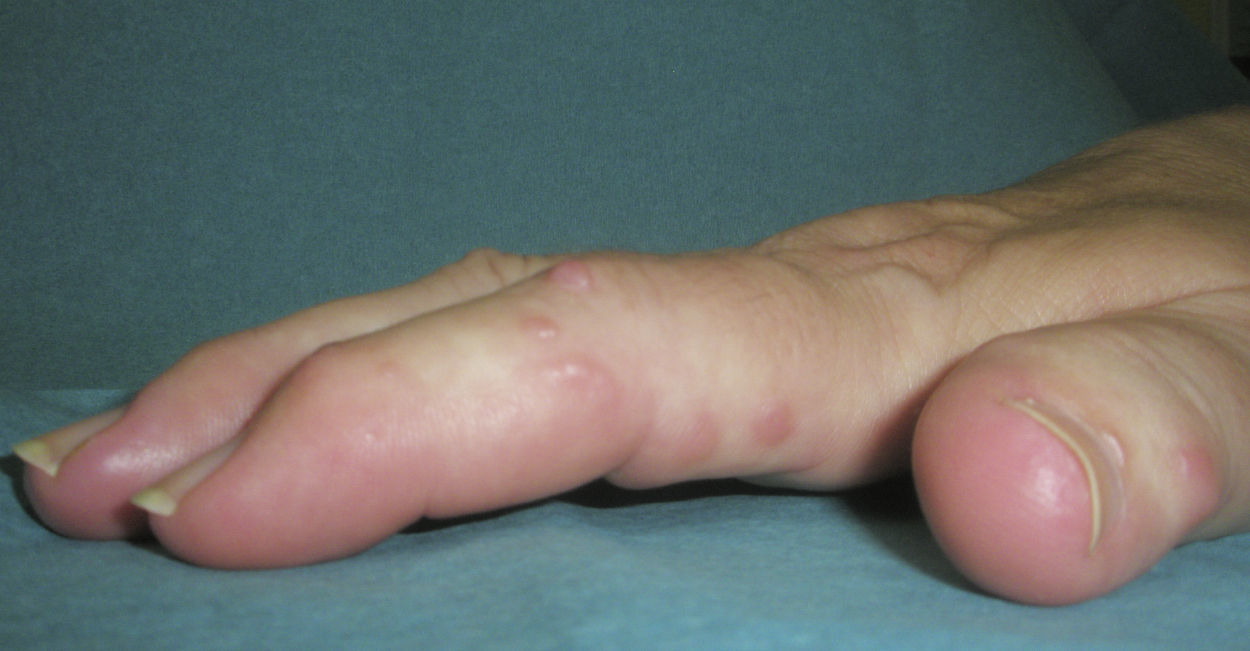
Manifestaciones clínicasSe caracteriza por: a) la asociación de poliartritis erosiva rápidamente progresiva, con daño y pérdida de la función articular, que conlleva la aparición de contractura de la musculatura flexora de los dedos (denominada por los autores franceses como main an griffe8); la artropatía erosiva severa ha sido descrita en varias ocasiones, pero es menos común26; b) nódulos cutáneos duros, lisos, no dolorosos y de curso intermitente, y c) esclerodactilia (figs. 1 y 2).


Los nódulos cutáneos se presentan normalmente en la cara dorsal de las manos, sobre todo a nivel de las metacarpofalanges y de las interfalanges proximales, muñecas, codos y rodillas, pero también han sido descritos en la nariz3, en las orejas8,27, en los hombros7, en la espalda17 y en el cuello17,28. Su tamaño oscila entre 2 y 20mm, su aspecto es rojizo y brillante, no son dolorosos, son móviles y de consistencia dura, y no dejan lesiones residuales tras su desaparición, que suele ocurrir entre los 6 y los 24 meses, en algunas ocasiones de forma espontánea2,21.
El fenómeno de Raynaud y la esclerodactilia pueden estar presentes en una proporción estimada del 35 y del 76%, respectivamente2. Por otro lado, en algunos casos se ha informado de frialdad digital de manos16 y rigidez matutina de hasta más de una hora29,30. La afectación de grandes articulaciones es poco frecuente, aunque puede ocurrir4.
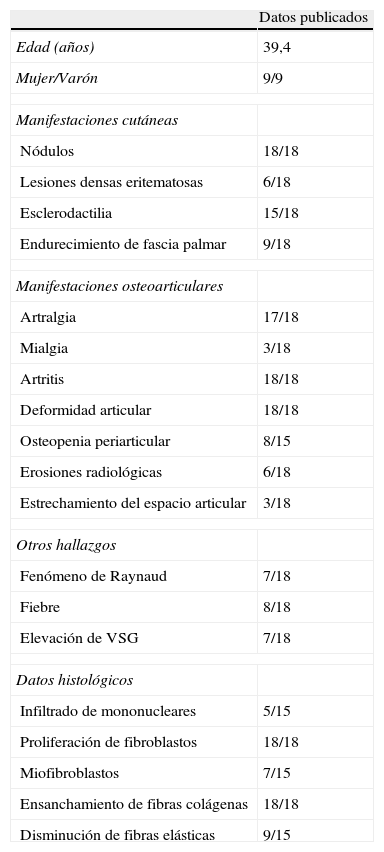
La fiebre suele acompañar a la sintomatología en el 40% de los casos8,31,32, y las manifestaciones sistémicas son muy poco comunes (tabla 1)16.
Características de los casos publicados de reumatismo fibroblástico
| Datos publicados | |
| Edad (años) | 39,4 |
| Mujer/Varón | 9/9 |
| Manifestaciones cutáneas | |
| Nódulos | 18/18 |
| Lesiones densas eritematosas | 6/18 |
| Esclerodactilia | 15/18 |
| Endurecimiento de fascia palmar | 9/18 |
| Manifestaciones osteoarticulares | |
| Artralgia | 17/18 |
| Mialgia | 3/18 |
| Artritis | 18/18 |
| Deformidad articular | 18/18 |
| Osteopenia periarticular | 8/15 |
| Erosiones radiológicas | 6/18 |
| Estrechamiento del espacio articular | 3/18 |
| Otros hallazgos | |
| Fenómeno de Raynaud | 7/18 |
| Fiebre | 8/18 |
| Elevación de VSG | 7/18 |
| Datos histológicos | |
| Infiltrado de mononucleares | 5/15 |
| Proliferación de fibroblastos | 18/18 |
| Miofibroblastos | 7/15 |
| Ensanchamiento de fibras colágenas | 18/18 |
| Disminución de fibras elásticas | 9/15 |
VSG: velocidad de sedimentación globular.
De Fam et al.1.
El diagnóstico se fundamenta en el estudio anatomopatológico, con una histología y una inmunohistoquímica características que sugieren que el RF podría estar incluido en las denominadas fibromatosis7,20. Se trata de un trastorno limitado a la dermis y al tejido celular subcutáneo, de tal modo que la epidermis, la aponeurosis y las estructuras musculares quedan exentas20. El examen histopatológico revela proliferación de fibroblastos y miofibroblastos, infiltrado mononuclear, incremento del estroma del colágeno, ensanchamiento marcado de la dermis y pérdida de fibras elásticas2–4,24,29,33. Estas fibras elásticas pueden estar presentes, y entonces aparecen comprimidas en una disposición en paralelo, similar a los cambios descritos para los queloides y la esclerodermia34–36. Alrededor de los vasos superficiales adyacentes al plexo capilar aparecen un número escaso de linfocitos20,21. En unos pocos casos se han estudiado biopsias sinoviales, observándose hiperplasia y proliferación de fibroblastos, tejido de granulación vascular y fibrosis, con sinoviocitos normales22,27,37.
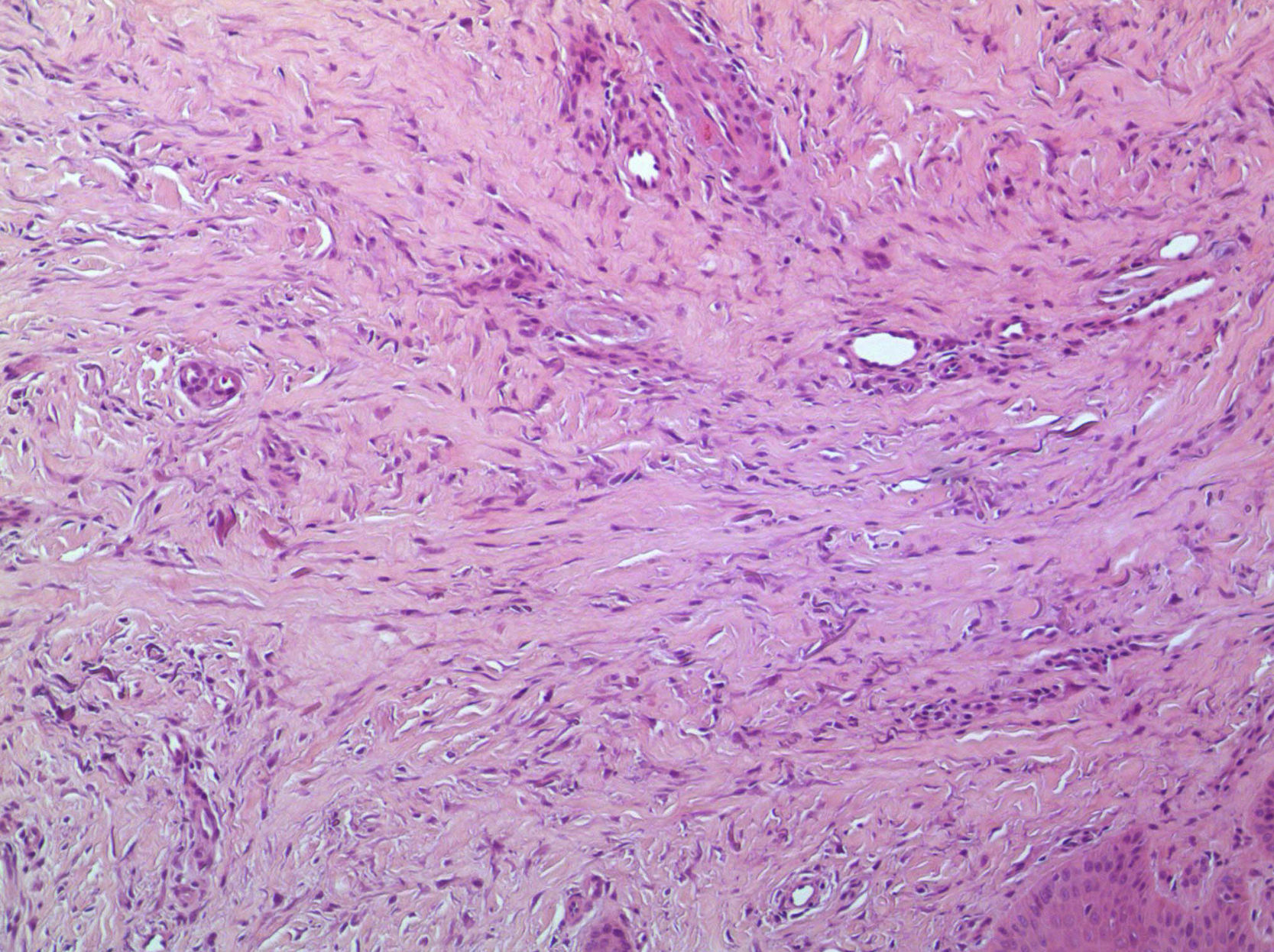
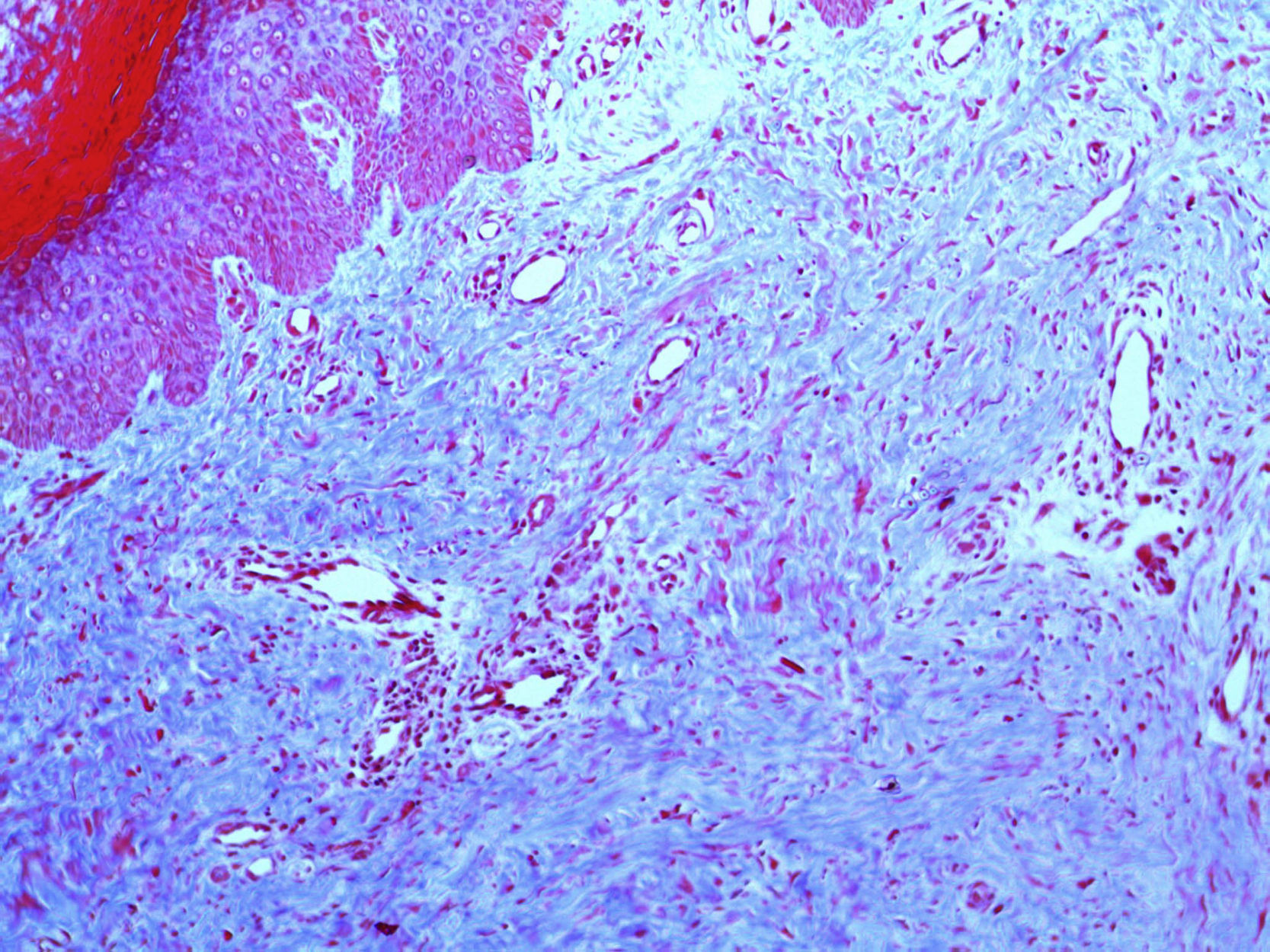
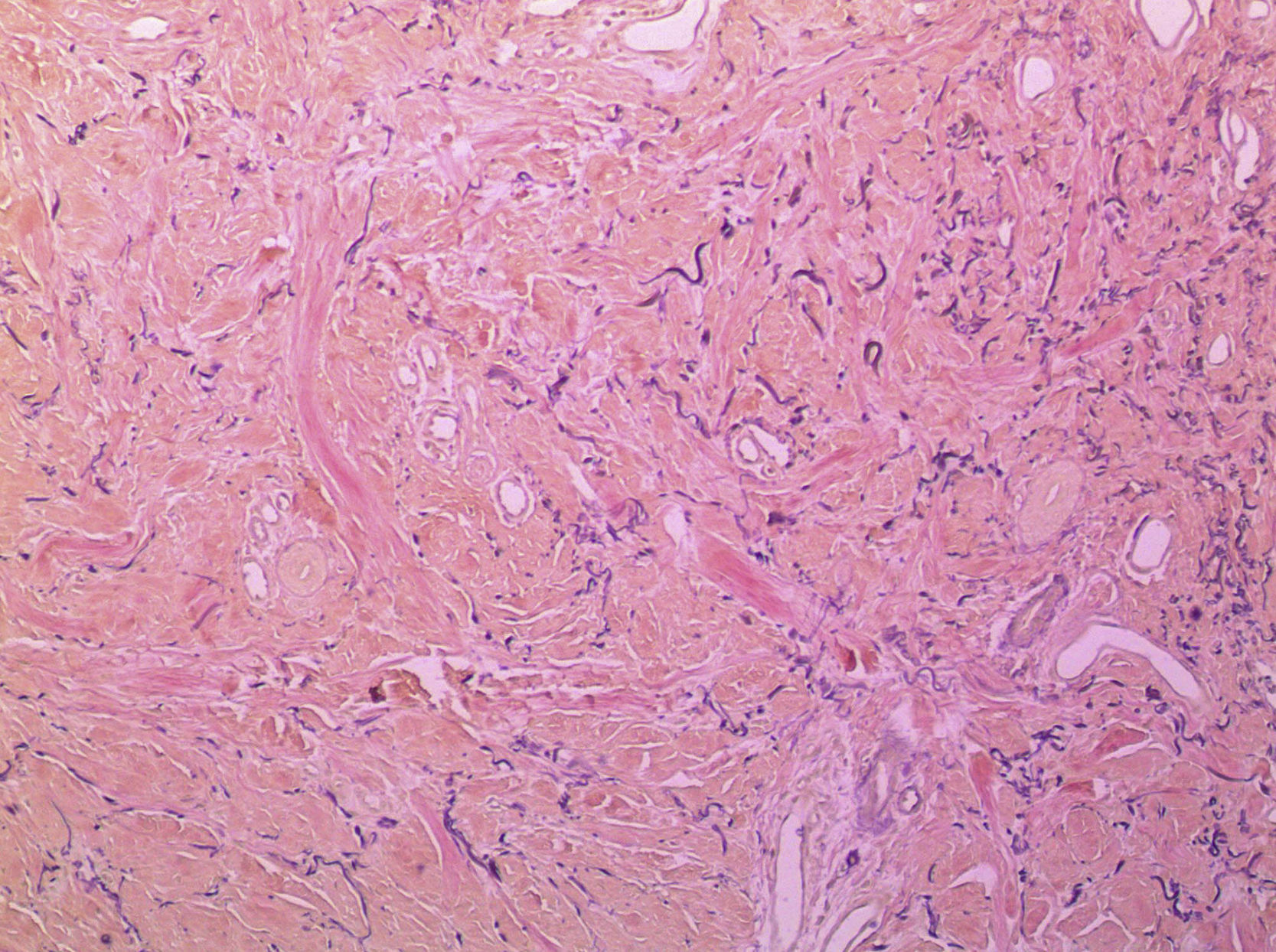
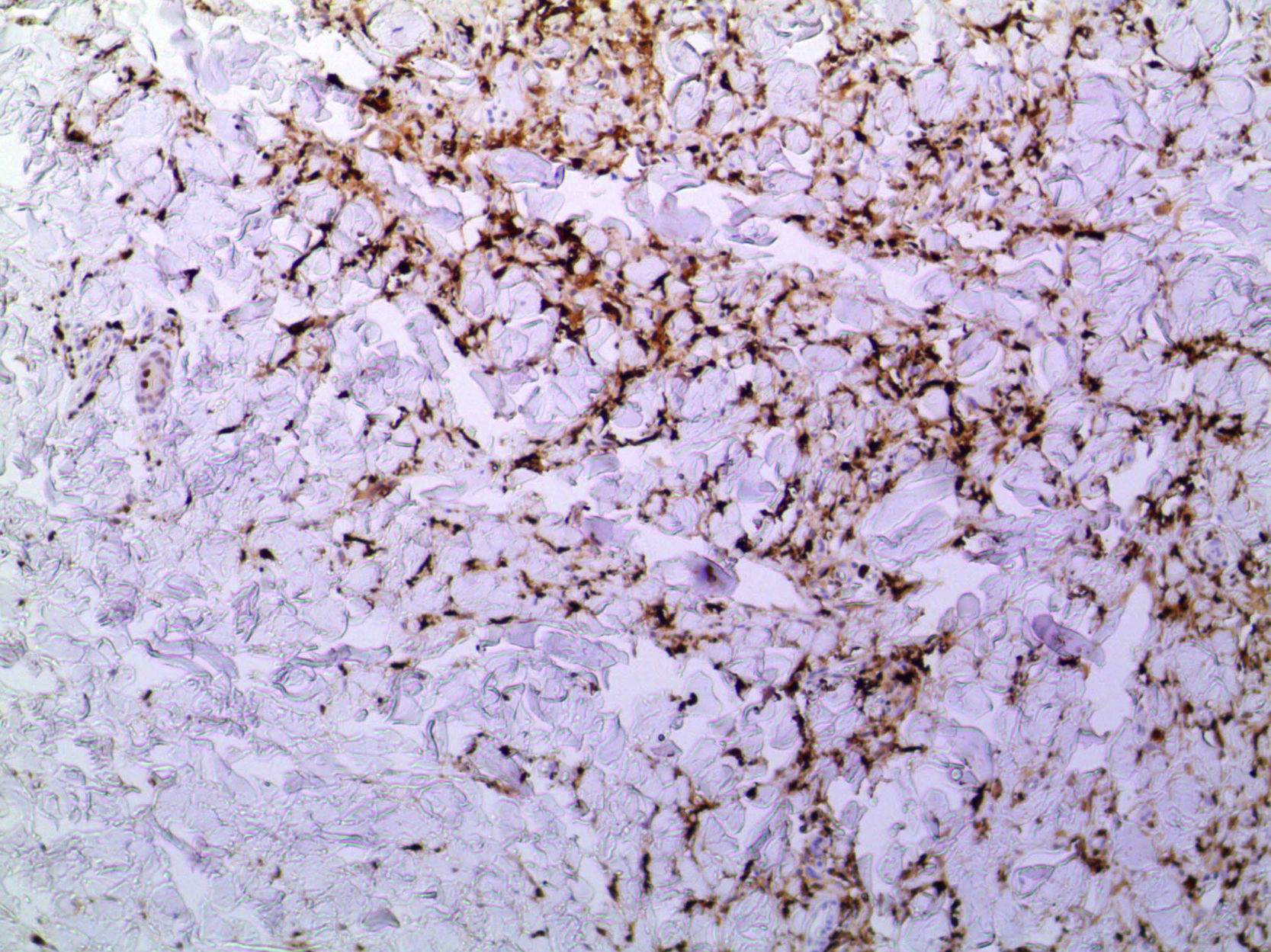
En la figura 3 se observa la proliferación de fascículos colágenos a nivel de la dermis con patrón irregular, leve infiltrado inflamatorio crónico perivascular, fragmentación y disminución de la cantidad de fibras elásticas, con presencia de abundantes células de citoplasma eosinófilo angulado y núcleos ovoideos de aspecto miofibrilar. En la tinción con tricrómico se identifica aumento del colágeno, lo que confirma su naturaleza fibroblástica (fig. 4). La tinción con solución de Van Gieson confirma la disminución de fibras elásticas (fig. 5). Inmunohistoquímicamente, estas células proliferantes son positivas para desmina, actina de músculo liso y anticuerpo monoclonal HHF-35, y negativas para CD1a, CD 34, CD 68, factor XIIIa y proteína S-100 (fig. 6)20,38.




La evolución de estas lesiones discurre en dos fases: a) fase aguda con infiltración de macrófagos y linfocitos, abundante proliferación de fibroblastos y miofibroblastos, engrosamiento y desorganización de fibras colágenas y pérdida de fibras elásticas, y b) fase crónica con escaso infiltrado de células mononucleares, proliferación más moderada de fibroblastos y miofibroblastos, engrosamiento de las fibras colágenas e intensa fibrosis dérmica1,4,17.
El diagnóstico se basa, por tanto, en la sospecha clínica y la confirmación anatomopatológica. Esto es así porque los controles analíticos no ayudan al diagnóstico, y las pruebas de imágenes —como la radiografía simple, la resonancia magnética nuclear (RMN) o la ecografía— suelen ser normales o bien muestran erosiones en la superficie articular, dependiendo de la evolución del cuadro.
Las erosiones detectadas por radiología simple aparecen en casos avanzados y localizadas en pequeñas articulaciones de manos y pies (metacarpofalángicas, metatarsofalángicas e interfalángicas), acompañándose de osteopenia periarticular y estrechamiento del espacio articular2,29,30,33. Existen casos de erosiones en huesos del carpo —como el trapecio— y acroosteólisis de falanges distales29. No obstante, las publicaciones no ofrecen demasiada información en cuanto a estas lesiones, y sobre todo en cuanto a los cambios radiológicos evolutivos (tabla 2)29.
Principales patrones radiológicos observados en función de tiempo evolutivo en 20 pacientes con reumatismo fibroblástico
| Patrones radiológicos | Meses | ||
| < 4 | ≥ 4 y < 12 | ≥ 12 | |
| Normal | 10/15 | 1/5 | 1/13 |
| Anormal | 5/15 | 4/5 | 12/13 |
| Aumento de partes blandas | 3 | 0 | 0 |
| Desmineralización | 2 | 2 | 2 |
| Erosión con espacio articular normal | 0 | 1 | 4 |
| Erosión con disminución del espacio articular | 0 | 1 | 6 |
| Sin información sobre patrón radiológico | 7 | 17 | 9 |
De Rety et al.29.
Solo la RMN ayudaría a detectar erosiones óseas de forma temprana, junto con cambios incipientes de tejido blando, además de ayudar a monitorizar la actividad de la enfermedad y su progresión4. De todos modos, el uso y la experiencia de la RMN aplicada a esta rara entidad son muy reducidos, y no parece que su aplicación se vaya a extender3,4. De hecho, bajo ninguna de estas técnicas se ha señalado alguna característica propia que la diferencie del resto de artropatías inflamatorias, tales como la artritis reumatoide.
Para el estudio de estos pacientes se han utilizado técnicas como la capilaroscopia, el test de función pulmonar, la tomografía axial computarizada (TAC) de tórax o la ecocardiografía transtorácica (ETT)30. Los resultados en ningún caso han sido concluyentes20.
En cuanto a los datos analíticos, los estudios bioquímicos, los datos de función hepática, renal y tiroidea, el hemograma, los reactantes de fase aguda (velocidad de sedimentación globular [VSG] y proteína C reactiva [PCR]), el análisis de orina, la serología general, el estudio de autoinmunidad (factor reumatoide [FR], anticuerpos antipéptido citrulinado [anti-CCP], anticuerpos antinucleares [ANA] y anticuerpos anticitoplasma de neutrófilos [ANCA]), el HLAB27 y las enzimas musculares son generalmente normales21,39.
Diagnóstico diferencialPor su gran similitud, la reticulohistiocitosis multicéntrica resalta entre las candidatas a la hora de establecer el diagnóstico diferencial del RF, sumando otras enfermedades tales como: artritis reumatoide, artritis psoriásica, osteoartritis erosiva, artritis gotosa, quiste mucinoso, dermatomiositis, enfermedad de Dupüytren, tumores/seudotumores, esclerodermia nodular, dermatofibromas, histiocitosis de células no-Langerhans, xantogranuloma, nódulo-artropatía-osteólisis (NAO), síndrome de Torg, histiocitosis nodular progresiva, mucinosis papular acral persistente, fibromatosis juvenil hialina, fibromatosis digital infantil y lipogranulomatosis de Farber20,37,40–42. Todas ellas asocian nódulos cutáneos y manifestaciones articulares (tablas 3 y 4).
Diagnóstico diferencial entre artropatías erosivas de manos, incluyendo reticulohistiocitosis multicéntrica
| IFP | Afectación simétrica | Erosiones | Osteoporosis | Espacio articular | |
| Reticulohistiocitosis multicéntrica | Presente | + | Marginal | - | Ampliado |
| Artritis reumatoide | Ausente | + | Marginal | + | Disminuido |
| Artritis psoriásica | Presente | – | Marginal con formación ósea perióstica | – | Ampliado/Disminuido |
| Osteoartritis erosiva | Presente | ± | Central | – | Disminuido |
| Artritis gotosa | Presente | ± | Marginal | – | Preservado/Disminuido |
| Reumatismo fibroblástico | Presente | + | Marginal/Central | + | Disminuido |
IFP: interfalángicas proximales.
De Trotta et al.10.
Principales diferencias entre RHM, FJH, Torg/NAO y RF
| RHM | Torg/NAO | FHJ | RF | |
| Media de años (comienzo de los síntomas) | 40 | 3,4 | 5 | 36 |
| Afectación cutánea | ||||
| Esclerodactilia | – | – | – | +/– |
| Radiología | ||||
| Osteólisis | – | + | + | +/– |
| Sinovitis | + | – | – | + |
| Biopsia | ||||
| Proliferación fibroblástica | – | + | + | + |
| Ensanchamiento de fibras colágenas | – | + | + | + |
| Inmunohistoquímica | CD68, FXIII+ | Sin información | PAS+ | Vimentina, α-SMA+ |
| Mutación genética | Sin información | MMP-2 | CMG-2 | Sin información |
RHM: reticulohistiocitosis multicéntrica; NAO: síndrome nodulosis, artropatía y osteolisis; FHJ: fibromatosis hialina juvenil; RF: reumatismo fibroblástico; α-SMA: smooth muscle actina; MMP-2: matrix metalloproteinase-2; CMG-2: capillary morphogenesis gene-2.
De Ji L. et al.25.
La reticulohistiocitosis multicéntrica, caracterizada por poliartritis severa, rigidez de manos y nódulos cutáneos, se distinguirse del RF por la presencia de células histiocíticas (histiocitos mononucleares), células gigantes multinucleadas (con diámetro de 50-100μm) con citoplasma eosinofílico y gránulos finos con apariencia de vidrio esmerilado, sin proliferación de fibroblastos y miofibroblastos10,11,43,44. La artritis reumatoide presenta los llamados nódulos reumatoideos, aunque la clínica, la radiología y las características serológicas nos permiten distinguirla fácilmente del RF33,45.
De igual modo ocurre con la dermatomiositis, que cursa con las denominadas pápulas o nódulos de Grotton que podrían confundirse con los nódulos del RF, aunque estos nódulos son más violáceos y suelen aparecer sobre prominencias óseas (nudillos, codos y rodillas). Su histología sería igualmente diferente, con mayor componente inflamatorio. La clínica de la dermatomiositis estaría más relacionada con un cuadro de debilidad muscular proximal en la cintura pelviana y escapular, y su asociación con otras manifestaciones clínicas, como el eritema periocular en heliotropo, hace que sea una entidad relativamente fácil de distinguir46.
La esclerodermia nodular es una forma particular y muy poco frecuente de esclerosis sistémica con múltiples nódulos queloideos. Ambas entidades se asocian a esclerodactilia y nódulos cutáneos, aunque nuevamente la histología ayuda a distinguirla, ya que en la esclerodermia nodular aparecen células queloideas sin proliferación fibroblástica e implican a glándulas exocrinas21,47.
La mucinosis papular acral persistente no suele asociarse a afectación articular, y los fibroblastos suelen estar rodeados de abundante contenido mucinoso40.
En niños, la fibromatosis juvenil hialina (OMIM 228600) es el principal diagnóstico sobre el que cabe enfrentar el RF48. Se trata de una rara enfermedad de herencia autosómica recesiva debido a mutaciones en el gen-2 de la morfogénesis capilar (CMG2). Estos niños también presentan contractura de la musculatura flexora de los dedos, erosiones óseas y nódulos cutáneos que imitan al RF. Sin embargo, existen algunos datos clínicos que lo diferencian: comienzo preferible en la infancia (principalmente en torno a los 5 años de edad); las lesiones cutáneas suelen aparecer en el tronco; hipertrofia gingival, y ausencia de esclerodactilia. Además, los hallazgos histológicos revelan la asociación de proliferación de fibroblastos y miofibroblastos junto con un característico depósito de sustancia hialina PAS-positiva amorfa48,49. La fibromatosis digital infantil es una enfermedad congénita o puede aparecer en niños antes de los 3 años de edad. Se caracteriza por múltiples nódulos que afectan a los dedos, aunque sin esclerodactilia ni compromiso articular. Su histología se caracteriza por una proliferación celular con infiltrado eosinofílico y cuerpos de inclusión paranuclear en el citoplasma50.
Finalmente, otras dos raras entidades pueden asociarse a nódulos subcutáneos y deformidad articular. La lipogranulomatosis de Farber (OMIM 228000) es una rara enfermedad de herencia autosómica recesiva perteneciente a las enfermedades por depósito lisosomal, causada por la deficiencia en la enzima ceramidasa ácida, con la consiguiente acumulación de ceramidasa en varios tejidos51,52. En el examen histológico los nódulos son muy diferentes respecto al RF, ya que contienen granulomas e inclusiones citoplasmáticas lipídicas dentro de macrófagos. El síndrome de Winchester (OMIM 277950) pertenece a los síndromes osteolíticos. Histológicamente los nódulos muestran proliferación difusa de fibroblastos en las capas más profundas de la dermis, con extensión hacia el tejido subcutáneo e infiltración linfocítica perivascular53.
PronósticoLos nódulos del RF no suelen dejar lesiones residuales tras su desaparición, que suele ocurrir entre 6 y 24 meses, a veces incluso de forma espontánea21. Cuando las lesiones que ocasiona el RF son diagnosticadas y tratadas de forma precoz, el proceso suele resolverse en el plazo de 2 a 8 meses, sin repercusiones funcionales en la vida cotidiana y laboral4,54.
Solo en los casos más evolucionados pueden originarse contractura de la musculatura flexora de los dedos (main an griffe8) y afectación articular rápidamente progresiva, con limitación y dolor a lo largo del balance articular.
TratamientoEl tratamiento no está consensuado. Con el uso de antiinflamatorios no esteroideos (AINE)23,55, corticoides5,8,17,55, metotrexato5,6, hidroxicloroquina4,54, leflunomida7, interferón alfa7,56, ciclofosfamida, colchicina27 y penicilamina16 se han obtenido resultados muy variados4,16,38.
La combinación de corticoides y metotrexato es la que cuenta con mayor experiencia en la actualidad y la que mejores resultados ha obtenido6. En general suele utilizarse la combinación de prednisona 20mg/día y metotrexato 2,5mg (10-15mg semanales), siempre acompañado de ácido fólico20,38. Algunos autores apuntan que los esteroides pueden estar implicados en la supresión de la proliferación de citoquinas fibróticas con la subsiguiente limitación en la activación de fibroblastos4. Así mismo, el metotrexato ha demostrado eficacia en la inhibición de la proliferación de fibroblastos en la sinovial17.
La hidroxicloroquina (200mg/12h) también ha sido frecuentemente utilizada en estos casos4,54. Su uso se fundamenta en la intervención sobre los lisosomas, dado que se ha observado un aumento de la proliferación lisosomal en los casos de RF54. Se conoce que estos agentes antimaláricos son bases débiles que aumentan el pH dentro de los lisosomas y afectan su actividad enzimática dependiente del equilibrio ácido-base citoplasmático. Además, estos fármacos son capaces de inhibir directamente la fosfolipasa lisosomal A y C57,58.
En casos refractarios se ha realizado tratamiento con anti-factor de necrosis tumoral alfa (anti-TNFα), concretamente con infliximab en dosis de 3mg/kg, en combinación con metotrexato, consiguiendo reducir los nódulos cutáneos con las tres primeras dosis (0, 2 y 6 semanas)26. La molécula de TNF-α es una citoquina proinflamatoria liberada por macrófagos, monocitos y linfocitos T, así como células específicas como las células dendríticas y los queratinocitos. Es conocida su participación en la activación del endotelio vascular, en la regulación de la respuesta inmune y en el metabolismo del tejido conectivo, gracias a la modulación de la función fibroblástica59; de ahí que su uso experimental en el RF se fundamente en esta última premisa. Así pues, el desarrollo de nuevos agentes biológicos aporta nuevas alternativas terapéuticas. En la experimentación animal, el interferón gamma (IFN-γ) inhibe la producción de IL-1β-induced MMP en los fibroblastos de la sinovial y protege del daño articular en las artritis precoces60. El antagonista TGF-β está implicado en la inhibición de la transformación de fibroblastos en miofibroblastos, utilizándose por ello en la reducción del depósito de matriz extracelular y las cicatrices61.
En no pocos casos al tratamiento médico se asoció tratamiento rehabilitador, con lo que mejoró la funcionalidad manual y desaparecieron casi por completo las dificultades ocasionadas de cara a la manipulación de objetos26,38. El tratamiento quirúrgico solo se ha empleado para corregir las deformidades digitales refractarias al tratamiento médico/rehabilitador20.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.