




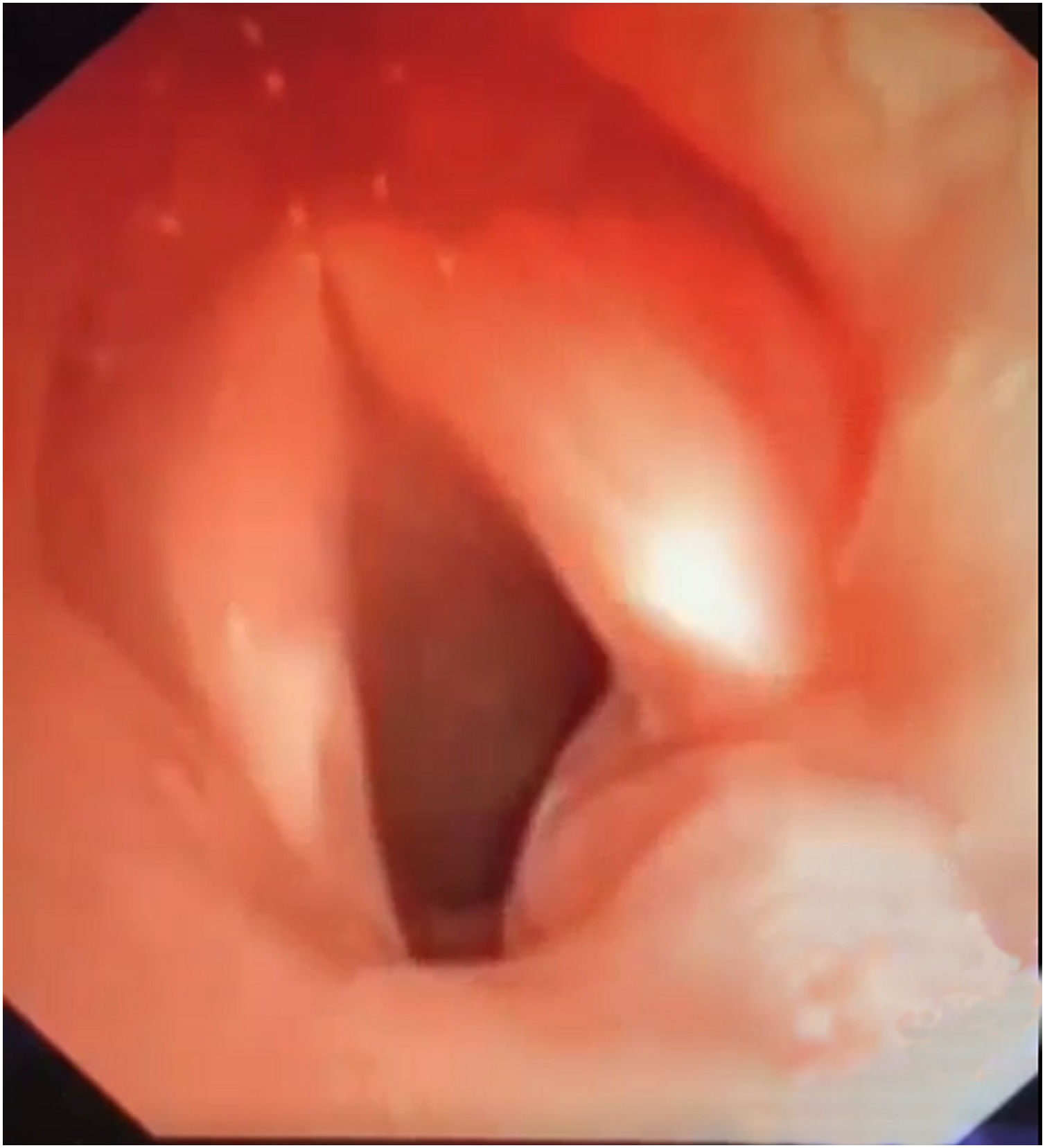
We present the case of a 4-year-old patient with stridor and severe difficulty in breathing, who required admission to a Paediatric ICU and invasive ventilation. Bronchoscopy examination revealed a type 2 laryngeal image according to the Benjamin Inglis classification (Fig. 1) (Appendix A, Video 1 of additional material). Endoscopic correction was performed with reconstruction of the cleft by suture in both planes (oesophageal and laryngotracheal), with interposition of fascial muscle lamina from the sternocleidomastoid muscle between both lines of suture. A repeat intervention was required after 15 days due to a new episode of stridor and respiratory difficulty due to obstruction of the airway by redundant inter-arytenoid tissue. This was resolved by diode laser photocoagulation of the oesophageal mucus swelling, with good subsequent evolution.

The following aspects stand out within the clinical history of our patient: diagnosis at one month of life of laryngotracheomalacia by nasofibroscopy, without other complications. There were also hypospadias, cryptorchidy and umbilical hernia. He was diagnosed persistent arterial ductus, which closed spontaneously at one year of life, and he required monitoring by the Neuropaediatric Unit due to hypotonia at breast feeding age, as well as having a peculiar phenotype with hypertelorism and left ptosis. Given all of the said factors, and following the diagnosis of laryngeal cleft, the suspicion of Opitz G/BBB syndrome was established, and it was confirmed by the finding of the hemizygotic c.346A >T mutation in the MID1 gene.
DiscussionLaryngeal clefts (LC) are rare congenital anomalies of the airways and digestive tract, with an incidence of 1/10,000–20,000 live new births, with a slight predominance of the male sex.1,2 They consist of the incomplete separation between the oesophagus and the airway from the start of the larynx.3
LC is a rare pathology that is caused by an incomplete formation of the tracheoesophageal septum during embryonic development. Depending on the severity of this defect, different anomalies may be produced such as an isolated laryngeal cleft, a tracheoesophageal fistula or oesophageal atresia.2
Clinical manifestations vary depending on the depth of the LC.4 There are several different classifications of laryngeal clefts, of which the most widely used is the one by Benjamin-Inglis,2,5 which differentiates 4 types of laryngeal cleft according to their extension. Types 1 and 2 usually present with mild respiratory symptoms and difficulty in eating, with frequent aspirations.1,3 It is striking that the patient in the case described, with type 2 LC, had no respiratory symptoms during the first years of life, and commenced with an episode of severe respiratory difficulty at the age of 4 years. Types 3 and 4 are more severe forms, with a more extensive communication between the oesophagus and the trachea.6 Symptoms commence in these forms in the neonatal period, with severe respiratory alterations,1,4,6 and they generally require early bronchoscopy due to episodes of apnoea and hypoxia.4
The diagnostic method of choice is direct laryngoscopy.4 It is important to take this pathology into account in the differential diagnosis of children with stridor, recurring respiratory infections and alterations in deglutition.3,6 Bronchoscopy makes it possible to classify and describe the extension of the defect, and it was the key to diagnosis in the case described.
The treatment of LC is medical as well as surgical.7 In medical terms, it is fundamental to prevent pulmonary complications associated with aspiration episodes, ensuring appropriate nutrition and using thickeners when this is necessary.4,7 It is also important to treat other comorbidities (gastric-oesophageal reflux, bronchial hyper-reactivity or food allergies, etc.) which may affect the airway and digestive tract.3,4 Medical treatment is especially important in type 1 LC, given that some authors state that appropriate medical treatment in these cases may delay or avoid the need for surgical repair.1,4 For surgical treatment, the access approach will depend on the type of cleft to be treated. In types 1 and 2 endoscopic repair of the laryngeal defect is preferred, reserving open surgery for types 3 and 4, together with those cases in which endoscopic technique was unsuccessful or failed.1,4
Although the majority of LC cases are isolated, they may also be associated with other congenital alterations, of which the most common are midline defects (cardiac, genital-urinary or gastrointestinal anomalies).6 The presence of a LC in association with other defects should make us consider certain specific syndromes, as it has been associated with the CHARGE, Pallister-Hall, VACTERL and Opitz G/BBB syndromes, as it was in the case described.1,2,4
Given that our patient had several congenital anomalies associated with LC, a genetic study was undertaken which finally confirmed the diagnosis of Opitz-G/BBB syndrome. This syndrome was described for the first time by Opitz in 1969,8 and up to 30% of the patients affected have a LC.9 The majority of the anomalies associated with this syndrome also affect the midline (hypertelorism, hypospadias, cardiac alterations, cleft palate …) suggesting that the mutation mainly affects morphogenic processes at this level.9,10 There are two clinically indistinguishable genetic subtypes of the Opitz-G/BBB syndrome: one variant linked to chromosome X and another dominant autosomal variant.10 The first of these variants is the most common, even though its frequency in the population is extremely low (1/100,000 new-born babies), and it is associated with mutations in the MID1 (Xp22) gene.10 The c.346A >T mutation, which was found in our case, has not been described beforehand in databases. Nevertheless, and given its nature, it is considered to be pathological and able in itself to explain the Opitz-G/BBB phenotype. As this syndrome is linked to chromosome X, only males develop this disease. Carrier women usually have hypertelorism as the sole manifestation, and the mother of the patient had this characteristic. However, genetic diagnosis of the parents was not performed as the family refused this.
To conclude, we should underline that LC are a rare anomaly which requires a high level of suspicion for diagnosis, especially in types 1 and 2, as they have symptoms which may often be confused with other diseases. Moreover, given that this pathology is often associated with certain syndromes, a genetic study should be considered in patients with other associated anomalies.
Authorship /collaboratorsAll of the authors express their agreement with the work produced and state that they actively collaborated in preparing the same.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: García-Muro C, Sáenz-Moreno I, Gómez-Sáez F, Navazo-Eguia A. Hendidura laríngea tipo 2 asociada a síndrome de Opitz G/BBB. Acta Otorrinolaringol Esp. 2022;73:196–198.