



As well as increased susceptibility to infections, autoimmune and inflammatory manifestations also eventuate due to dysregulation of immune system in a substantial proportion of patients with primary immunodeficiency (PID). Autoimmune and inflammatory manifestations can occur prior or after diagnosis of PID. This study aimed to evaluate autoimmune and inflammatory complications among all types of PID patients in childhood and to emphasize the importance of these findings as a warning sign to diagnose PIDs.
MethodsMedical records of 1036 patients with PID, followed up between 2003 and 2019, were retrospectively screened for occurrence of autoimmunity and inflammation. During this time, demographic features, autoimmune/inflammatory findings and initial time, genetic mutations, laboratory and clinical follow up findings, treatment regimens and outcomes were recorded.
ResultsAutoimmune and inflammatory manifestations were observed in 83 patients (10.1%). The median age of autoimmunity initial time was 61.3±53 months. Sixty-seven (80.7%) patients presented with autoimmune and inflammatory manifestations, and these findings had occurred during 16 patients’ (19.3%) follow-up. The most common autoimmune manifestations were autoimmune hematologic (51.8%) and endocrine diseases (26.5%). Fifty patients (60.2%) had a single autoimmune/inflammatory manifestation, however 23 patients (27.7%) had two, eight patients (9.6%) had three and two patients (2.4%) had four different types of autoimmune/inflammatory manifestations. The frequency of autoimmune and inflammatory manifestations in phagocyte defects (56%), combined immune deficiencies (53%) and immune dysregulation diseases (52%) were observed higher than other forms of PIDs. During follow-up 13 (15.7%) patients died.
ConclusionAutoimmune/inflammatory manifestations are associated with high morbidity in patients with PIDs and may precede the diagnosis of PID in childhood. Therefore, physicians must be aware of underlying possible immune deficiency and patients with known PIDs should be evaluated for autoimmune and inflammatory complications.
Primary immunodeficiencies are hereditary diseases that are described by impaired immune response.1 Excluding selective IgA deficiency, the prevalence of PID is reported at 1/10,000 in developed countries.1 It is considered that the prevalence of PID in our country is higher because of frequent consanguineous marriages. Primary immune deficiencies are especially characterized by susceptibility to infections. Autoimmunity/inflammation and malignancy are also other components of PIDs. The defects in central and peripheral regulatory systems of T and/or B cell development stages, antigen overload because of persistent and chronic infections, increased load of apoptotic cells and insufficiency of immune complexes clearance are mechanisms that caused appearing autoimmune and inflammatory findings.2–5 The reported frequencies of autoimmune/inflammatory manifestations in PIDs are quite variable according to the type of PIDs. Autoimmune/inflammatory manifestations can occur prior to or after the diagnosis of PID at any age. The first symptom in some PIDs can occur as autoimmunity. There is no limitation of any organ or tissue; autoimmune cytopenia, endocrinopathies, hepatitis, autoimmune lung disease, glomerulonephritis, or lupus can be observed. Autoimmune findings may also be accompanied by lymphoproliferation.
Hitherto, all reported studies on the frequency and distribution of autoimmune and inflammatory manifestations in patients with PIDs had been done in groups that mostly included adult patients. The primary aim of this study was to evaluate the autoimmune, inflammatory and lymphoproliferative complications among a large group of all types of PID patients in childhood. Secondly, we have intended to assess the importance of autoimmune and inflammatory manifestations as a warning sign for the diagnosis of PIDs in childhood.
Material and methodsData were retrospectively collected from the medical records of 1036 pediatric patients with PID followed by University of Health Sciences Izmir Dr. Behcet Uz Children's Education and Research Hospital, Division of Pediatric Allergy and Immunology, between 2003 and 2019. The diagnosis of the immunodeficiency was based on the diagnostic criteria of the European Society for Immunodeficiencies (ESID).6 Patients were then classified into different categories of PIDs in accordance with the International Union of Immunological Societies 2017 Primary Immunodeficiency Diseases Committee Report.7 Two hundred and fourteen patients with the diagnosis of transient hypogammaglobulinemia of infancy who made a full recovery during follow-up were excluded from the study. No autoimmunity/inflammation was detected in those cases. A total of 822 patients were included in the study. Our study group does not include patients with hereditary autoinflammatory diseases. Gender of patients, consanguinity, age of diagnosis, age of initial time of symptoms associated with PID, current age, genetic mutations, autoimmune and inflammatory findings, laboratory and clinical follow up findings, treatment regimens and outcomes were recorded. The initial time of autoimmune/inflammatory manifestations was established and compared with the time of PID diagnosis.
In our study the autoimmune/inflammatory manifestations were classified according to “World Health Organization: International statistical classification of diseases and related health problems Tenth Revision”.8,9 Then the autoimmune/inflammatory manifestations were grouped according to systems. Dermatitis, psoriasis, alopecia, lichen planus and vitiligo were included in dermatologic manifestations. Autoimmune neutropenia, autoimmune hemolytic anemia (AHA), immune thrombocytopenic purpura (ITP) and hemophagocytic lymphohistiocytosis (HLH) were included in hematologic manifestations. Autoimmune thyroiditis and Type 1 Diabetes mellitus (DM) were included in endocrinological manifestations. Pernicious anemia, colitis ulcerosa, celiac disease, autoimmune hepatitis, granulomatous hepatitis, inflammatory bowel disease, autoimmune enteropathy, stomatitis and recurrent aphthous mouth ulcers were included in gastrointestinal manifestations. Encephalitis, myasthenia gravis, multiple sclerosis, myopathy and Guillain–Barré syndrome were included in neurological manifestations; granulomatous lung disease was included in respiratory manifestations; and arthritis, juvenile rheumatoid arthritis and dermatomyositis were included in rheumatological manifestations.9 Non-infectious and non-malignant hepatosplenomegaly and lymphadenopathy was evaluated as lymphoproliferation.
This retrospective study was approved by the University of Health Sciences Izmir Dr. Behcet Uz Children's Education and Research Hospital Ethics Committee on 27.11.2017 (decision no 2017-167).
SPSS 11.0 (Statistical Package for Social Sciences) packet program was used for analyzing the study data. Descriptive statistics were expressed by numbers and percent. Numeric data was expressed by mean and standard deviations. Chi-square test was used to compare independent categorical variables. Mann-Whitney U test was performed to compare the autoimmunity age according to prognosis and p value <0.05 was considered statistically significant.
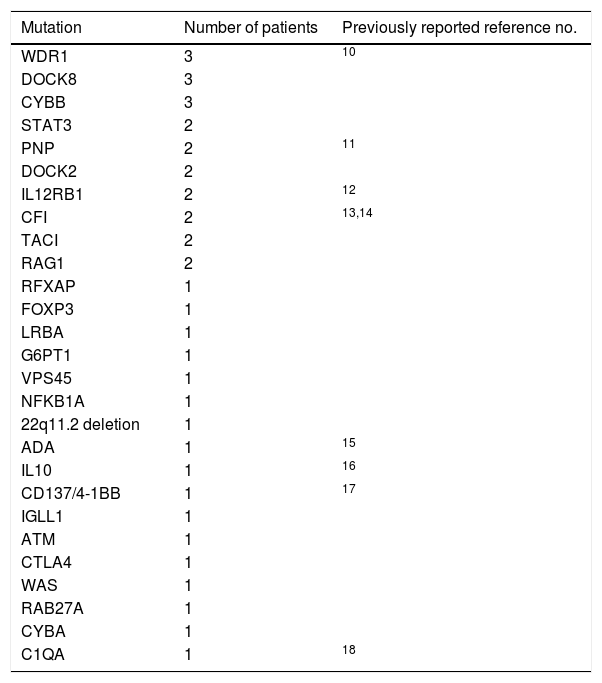
ResultsAutoimmune and inflammatory manifestations were observed in 10.1% (83 patients) of patients with PID. Forty-six (55.4%) of the patients were male. Consanguinity was present in the history of 29 patients (34.9%). The mean age of onset symptoms associated with PID was 49.2±49.5 months and the mean age of autoimmune/inflammatory manifestations’ initial time was 61.3±53 months. The mean age of diagnosis was 65±52.8 months. The current age of alive patients was 134.1±82.2 months. Age at death of patients who died during the follow-up was 84±68.6 months. Diagnosis was genetically confirmed in 40 (48.2%) patients (Table 1).
Distribution of determined mutations of PID patients with autoimmune/inflammatory manifestations.
| Mutation | Number of patients | Previously reported reference no. |
|---|---|---|
| WDR1 | 3 | 10 |
| DOCK8 | 3 | |
| CYBB | 3 | |
| STAT3 | 2 | |
| PNP | 2 | 11 |
| DOCK2 | 2 | |
| IL12RB1 | 2 | 12 |
| CFI | 2 | 13,14 |
| TACI | 2 | |
| RAG1 | 2 | |
| RFXAP | 1 | |
| FOXP3 | 1 | |
| LRBA | 1 | |
| G6PT1 | 1 | |
| VPS45 | 1 | |
| NFKB1A | 1 | |
| 22q11.2 deletion | 1 | |
| ADA | 1 | 15 |
| IL10 | 1 | 16 |
| CD137/4-1BB | 1 | 17 |
| IGLL1 | 1 | |
| ATM | 1 | |
| CTLA4 | 1 | |
| WAS | 1 | |
| RAB27A | 1 | |
| CYBA | 1 | |
| C1QA | 1 | 18 |
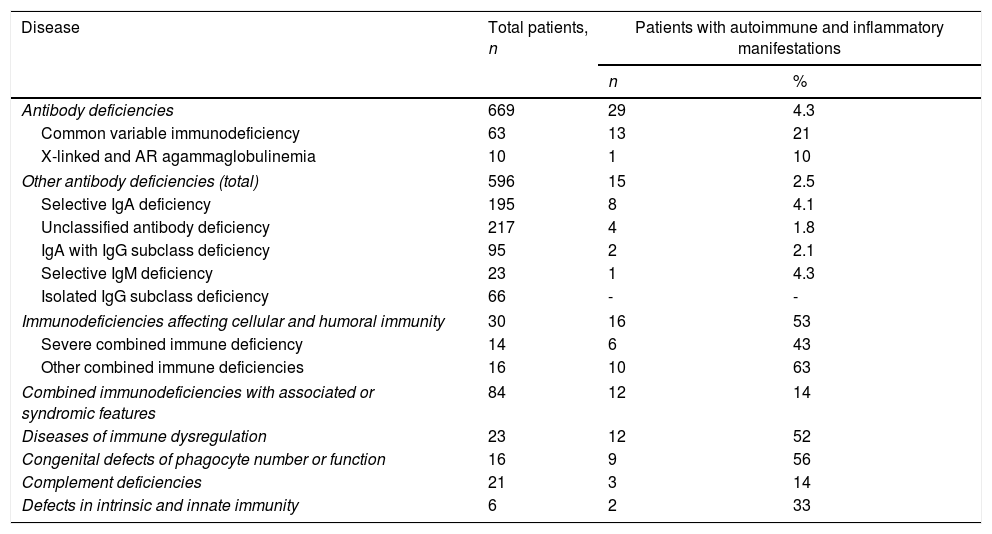
When the prevalence of autoimmunity was evaluated according to diagnostic groups, the frequency of autoimmune/inflammatory manifestations in defects of phagocyte (56%), combined immune deficiencies (53%) and diseases of immune dysregulation (52%) were observed higher than the other forms of PIDs (Table 2). Demographic features of the patients according to PID types are summarized in Table 3.
The prevalence of autoimmune and inflammatory findings in PID diagnostic groups.
| Disease | Total patients, n | Patients with autoimmune and inflammatory manifestations | |
|---|---|---|---|
| n | % | ||
| Antibody deficiencies | 669 | 29 | 4.3 |
| Common variable immunodeficiency | 63 | 13 | 21 |
| X-linked and AR agammaglobulinemia | 10 | 1 | 10 |
| Other antibody deficiencies (total) | 596 | 15 | 2.5 |
| Selective IgA deficiency | 195 | 8 | 4.1 |
| Unclassified antibody deficiency | 217 | 4 | 1.8 |
| IgA with IgG subclass deficiency | 95 | 2 | 2.1 |
| Selective IgM deficiency | 23 | 1 | 4.3 |
| Isolated IgG subclass deficiency | 66 | - | - |
| Immunodeficiencies affecting cellular and humoral immunity | 30 | 16 | 53 |
| Severe combined immune deficiency | 14 | 6 | 43 |
| Other combined immune deficiencies | 16 | 10 | 63 |
| Combined immunodeficiencies with associated or syndromic features | 84 | 12 | 14 |
| Diseases of immune dysregulation | 23 | 12 | 52 |
| Congenital defects of phagocyte number or function | 16 | 9 | 56 |
| Complement deficiencies | 21 | 3 | 14 |
| Defects in intrinsic and innate immunity | 6 | 2 | 33 |
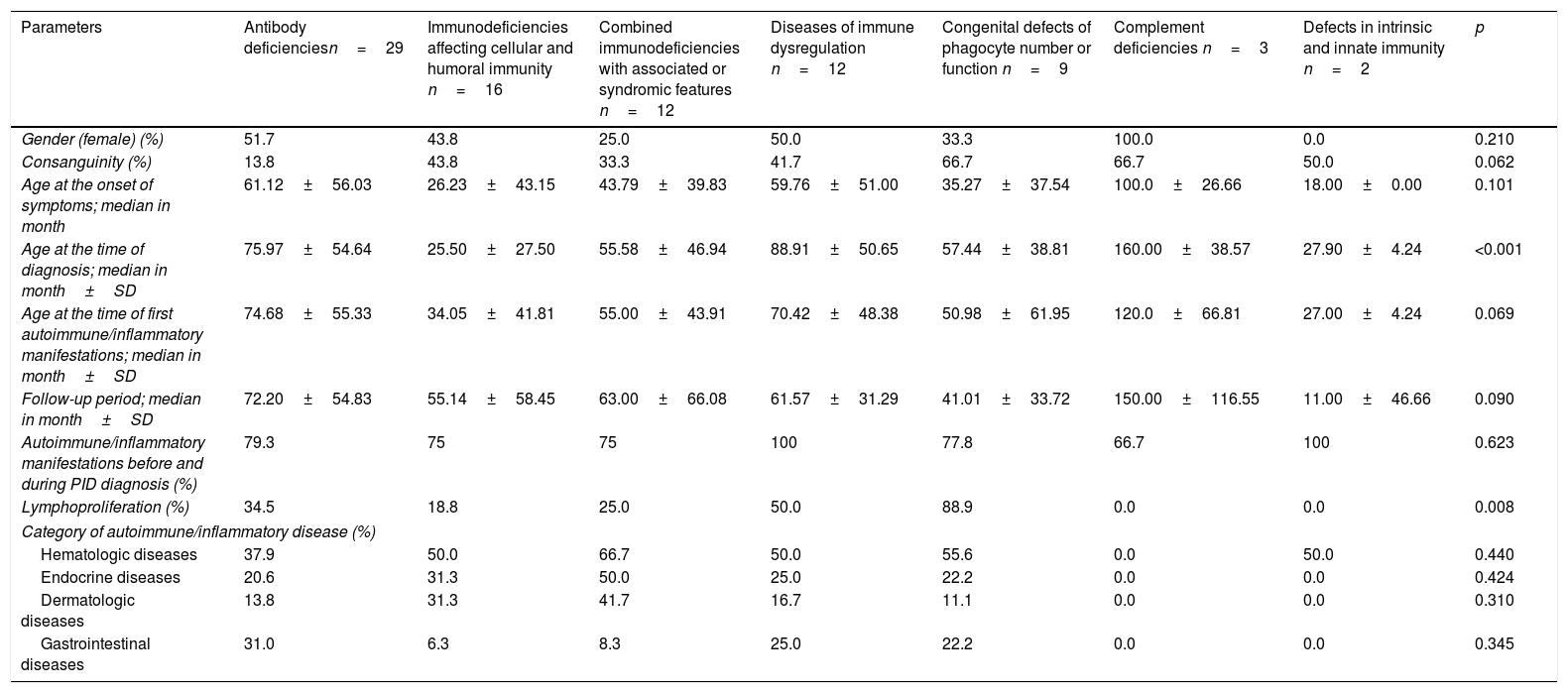
Demographic, clinical features and the frequency of most common autoimmune/inflammatory manifestations by PID type.
| Parameters | Antibody deficienciesn=29 | Immunodeficiencies affecting cellular and humoral immunity n=16 | Combined immunodeficiencies with associated or syndromic features n=12 | Diseases of immune dysregulation n=12 | Congenital defects of phagocyte number or function n=9 | Complement deficiencies n=3 | Defects in intrinsic and innate immunity n=2 | p |
|---|---|---|---|---|---|---|---|---|
| Gender (female) (%) | 51.7 | 43.8 | 25.0 | 50.0 | 33.3 | 100.0 | 0.0 | 0.210 |
| Consanguinity (%) | 13.8 | 43.8 | 33.3 | 41.7 | 66.7 | 66.7 | 50.0 | 0.062 |
| Age at the onset of symptoms; median in month | 61.12±56.03 | 26.23±43.15 | 43.79±39.83 | 59.76±51.00 | 35.27±37.54 | 100.0±26.66 | 18.00±0.00 | 0.101 |
| Age at the time of diagnosis; median in month±SD | 75.97±54.64 | 25.50±27.50 | 55.58±46.94 | 88.91±50.65 | 57.44±38.81 | 160.00±38.57 | 27.90±4.24 | <0.001 |
| Age at the time of first autoimmune/inflammatory manifestations; median in month±SD | 74.68±55.33 | 34.05±41.81 | 55.00±43.91 | 70.42±48.38 | 50.98±61.95 | 120.0±66.81 | 27.00±4.24 | 0.069 |
| Follow-up period; median in month±SD | 72.20±54.83 | 55.14±58.45 | 63.00±66.08 | 61.57±31.29 | 41.01±33.72 | 150.00±116.55 | 11.00±46.66 | 0.090 |
| Autoimmune/inflammatory manifestations before and during PID diagnosis (%) | 79.3 | 75 | 75 | 100 | 77.8 | 66.7 | 100 | 0.623 |
| Lymphoproliferation (%) | 34.5 | 18.8 | 25.0 | 50.0 | 88.9 | 0.0 | 0.0 | 0.008 |
| Category of autoimmune/inflammatory disease (%) | ||||||||
| Hematologic diseases | 37.9 | 50.0 | 66.7 | 50.0 | 55.6 | 0.0 | 50.0 | 0.440 |
| Endocrine diseases | 20.6 | 31.3 | 50.0 | 25.0 | 22.2 | 0.0 | 0.0 | 0.424 |
| Dermatologic diseases | 13.8 | 31.3 | 41.7 | 16.7 | 11.1 | 0.0 | 0.0 | 0.310 |
| Gastrointestinal diseases | 31.0 | 6.3 | 8.3 | 25.0 | 22.2 | 0.0 | 0.0 | 0.345 |
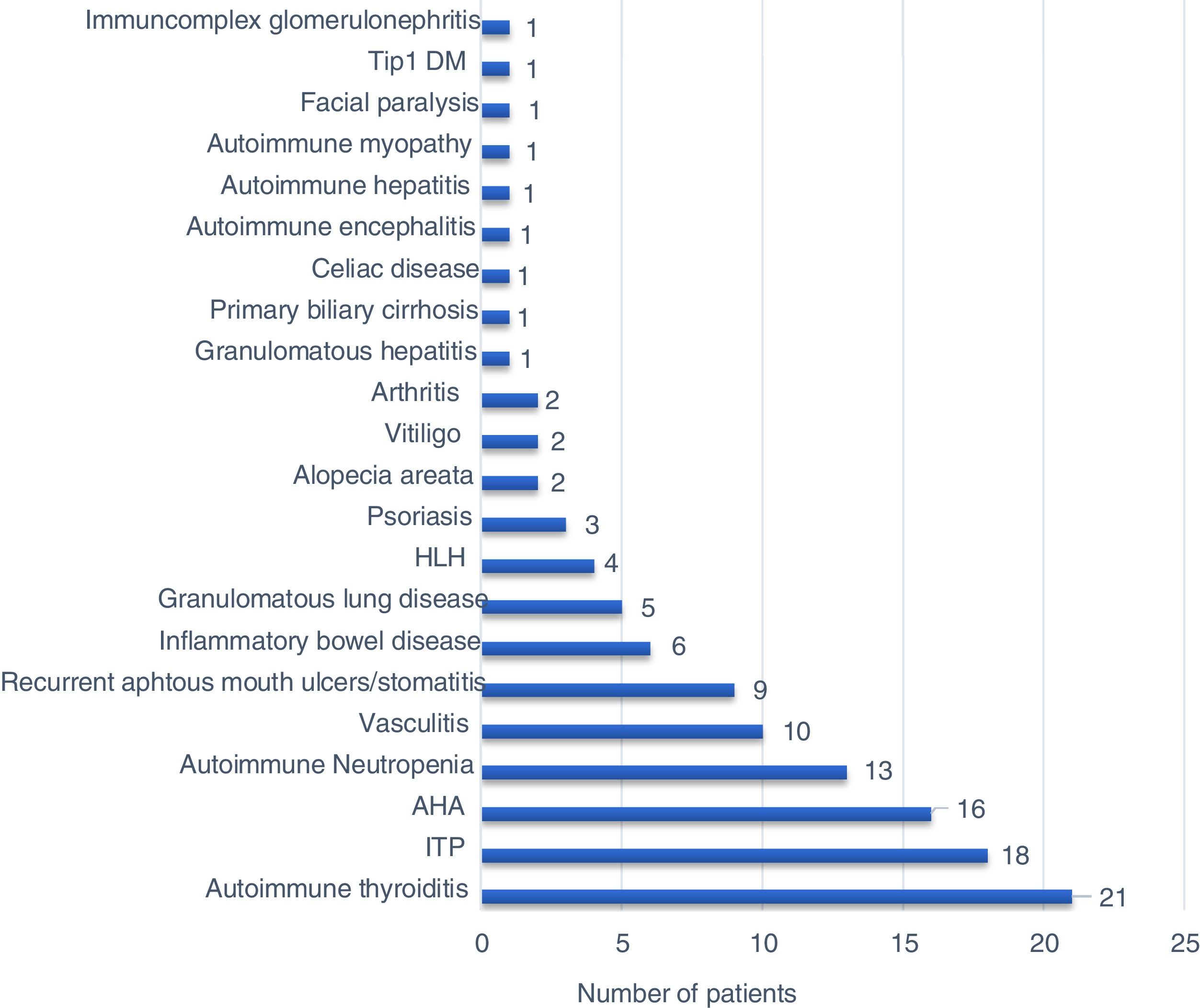
Sixty-seven (80.7%) patients presented with autoimmune/inflammatory manifestations, and these findings had occurred during 16 patients’ (19.3%) follow-up. There was no significant difference between PID types regarding the initial age of autoimmune/inflammatory manifestations. The percentages of the onset of autoimmune/inflammatory manifestations before and during PID diagnosis were also similar in all PID types (Table 3). The most common autoimmune/inflammatory manifestations were autoimmune thyroiditis (25.3%) and autoimmune thrombocytopenia (21.6%) (Fig. 1). When it was evaluated according to affected systems 51.8% hematological, 26.5% endocrinological, 20.5% dermatological, 19.3% gastrointestinal, 10.8% rheumatological 4.8% pulmonary and 2.4% neurological manifestations were identified. No significant difference was found regarding the affected system in PID types (Table 3).

Fifty-three (69.3%) patients had autoantibody positivity. Anti-T, anti-M, direct coombs and antinuclear antibody (ANA) were positive respectively in 24.5%, 26.4%, 30.1% and 3.75% of patients. Lymphoproliferation was detected in 36.1% (30 patients) of the cases with autoimmunity/inflammation. When lymphoproliferative manifestations were evaluated according to frequency; hepatosplenomegaly in 18 patients (21.7%), lymphadenopathy in 10 patients (12%) and splenomegaly in eight patients (9.6%) were determined. Lymphoproliferative findings were most common in phagocytic system defects (88.9%) and there was no lymphoproliferation in complement defects and innate immune system defects (Table 3). The mean age of initial time of autoimmunity was significantly higher in patients in whom lymphoproliferation occurred (79.5±54.2 vs 51.0±49.9, p=0.018).
Fifty patients (60.2%) had a single autoimmune/inflammatory manifestation, while 23 patients (27.7%) had two, eight patients (9.6%) had three and two patients (2.4%) had four different types of autoimmune/inflammatory manifestations. The patients were evaluated in terms of single and multiple autoimmunity/inflammation; a significantly higher rate of multiple autoimmune signs was observed in males. There was no significant difference according to the number of organs involved between the diagnostic groups. Significant multiple autoimmune manifestations were found in patients with endocrine and gastrointestinal system involvement (Table 4) (p=0.001).
Demographic and clinical features according to the number of autoimmune/inflammatory manifestations and outcomes.
| Single autoimmunity/inflammation(n=50) | Multiple autoimmunity/inflammation(n=33) | p-Value | Survivors (n=70) | Non-survivors (n=13) | p-Value | |
|---|---|---|---|---|---|---|
| Female sex (%) | 27 (73.0) | 10 (27.0) | 0.034 | 29 (78.4) | 8 (21.6) | 0.180 |
| Male sex (%) | 23 (50.0) | 23 (50.0) | 41 (89.1) | 5 (10.9) | ||
| Consanguinity (%) | ||||||
| No | 33 (61.1) | 21 (38.9) | 0.526 | 50 (92.6) | 4 (7.4) | 0.016 |
| First degree | 11 (52.4) | 10 (47.8) | 14 (66.7) | 7 (33.3) | ||
| Second and third degree | 6 (75.0) | 2 (25.0) | 6 (75.0) | 2 (25.0) | ||
| Age at the time of first symptoms; median in month±SD | 54.85±50.94 | 40.79±46.91 | 0.208 | 52.84±49.30 | 29.91±48.34 | 0.126 |
| Age at the time of diagnosis; median in month±SD | 72.90±53.46 | 53.06±50.45 | 0.095 | 68.08±51.56 | 48.46±58.91 | 0.221 |
| Age at the time of first autoimmune symptoms; median in month±SD | 68.57±56.48 | 50.31±46.03 | 0.125 | 64.45±52.63 | 44.40±54.18 | 0.213 |
| Follow-up period; median in month±SD | 63.09±52.73 | 71.87±63.57 | 0.498 | 69.68±58.27 | 41.86±41.34 | 0.170 |
| Lymphoproliferation (%) | ||||||
| No | 31 (58.5) | 22 (41.5) | 0.665 | 47 (88.7) | 6 (11.3) | 0.148 |
| Yes | 19 (63.3) | 11 (36.7) | 23 (76.7) | 7 (23.3) | ||
| PID category (%) | ||||||
| Antibody deficiencies | 22 (75.9) | 7 (24.1) | 0.353 | 26 (89.6) | 3 (10.3) | 0.198 |
| Immunodeficiencies affecting cellular and humoral immunity | 8 (50.0) | 8 (50.0) | 10 (62.5) | 6 (37.5) | ||
| Combined immunodeficiencies with associated or syndromic features | 5 (41.7) | 7 (58.3) | 11 (91.6) | 1 (8.3) | ||
| Diseases of immune dysregulation | 8 (66.7) | 4 (33.3) | 11 (91.6) | 1 (8.3) | ||
| Congenital defects of phagocyte number or function | 4 (44.4) | 5 (55.6) | 7 (77.8) | 2 (22.2) | ||
| Complement deficiencies | 2 (66.7) | 1 (33.3) | 3 (100.0) | 0 (0.0) | ||
| Defects in intrinsic and innate immunity | 1 (50.0) | 1 (50.0) | 2 (100.0) | 0 (0.0) | ||
| Category of autoimmune diseases | ||||||
| Hematologic diseases (%) | ||||||
| No | 32 (72.7) | 12 (27.3) | 0.01 | 39 (88.6) | 5 (11.4) | 0.252 |
| Yes | 18 (42.8) | 24 (57.2) | 31 (79.5) | 8 (20.5) | ||
| Endocrine diseases (%) | ||||||
| No | 43 (70.5) | 18 (29.5) | 0.001 | 50 (82.0%) | 11 (19.0%) | 0.323 |
| Yes | 7 (31.8) | 15 (68.2) | 21 (95.5%) | 1 (4.5%) | ||
| Gastrointestinal diseases (%) | ||||||
| No | 44 (65.7) | 23 (34.3) | 0.039 | 58 (86.6%) | 9 (13.4%) | 0.253 |
| Yes | 6 (37.5) | 0 (62.5) | 12 (75.0%) | 4 (25.0%) | ||
| Dermatologic diseases (%) | ||||||
| No | 42 (63.6) | 24 (36.4) | 0.213 | 56 (84.8) | 10 (15.2) | 0.114 |
| Yes | 8 (47.1) | 9 (52.9) | 17 (100.0) | 0 (0.0) | ||
| Autoimmune/inflammatory manifestation (%) | ||||||
| Single | 45 (90.0%) | 5 (10.0%) | 0.081 | |||
| Multiple | 25 (75.8%) | 8 (24.2%) | ||||
Parenteral immunoglobulin in 56 (67.4%) patients, glucocorticoids in 38 (45%) patients, azathioprine in two (2.4%) patients, sirolimus in one (1.2%) patient, tocilizumab in one (1.2%) patient and rituximab in one (1.2%) patient were performed for treatment of autoimmune/inflammatory manifestations. Levothyroxine in nine patients (10.8%), insulin replacement in one patient (1.2%), psoralen ultra-violet A (PUVA) in three patients (3.6%), interferon gamma in six patients (7.2%), interferon alpha in two patients (2.4%), and adenosine deaminase (ADA) enzyme replacement in one patient (1.2%) were also applied. Celiac diet was started for one patient, liver transplantation was performed for one patient and splenectomy was performed for another patient. Twelve patients underwent hematopoietic stem cell transplantation.
During follow-up, thirteen (15.7%) patients died. Six patients died because of sepsis (46.2%). The other causes of death were cardiac failure (n=1) in a patient with CVID and granulomatous lung disease, lymphoma (n=1) in a patient with ADA deficiency and development of hemophagocytic lymphohistiocytosis (n=1) in a patient with chronic granulomatous disease. Three of the 12 patients who received hematopoietic stem cell transplantation (HSCT) and one patient who underwent liver transplantation died. There was no significant difference according to gender, autoimmunity age, age of diagnosis, presence of single or multiple autoimmune manifestations, presence of lymphoproliferation, diagnosis, and organ involvement, except for a relatively higher rate of consanguinity in parents of the non-survivors (Table 4).
DiscussionThe frequencies of autoimmune/inflammatory manifestations related with PIDs are quite variable. In a study which included 247 adult and children with PID registered in the Slovenian national PID registry, the ratio of autoimmunity was 22%.19 In a retrospective study from patients registered in the French National Database, the ratio of autoimmune/inflammatory manifestations in patients with PID was reported as 26.2%. A total of 2163 adults and children (aged 0.5-92 years old) were included in that study.24 As mentioned above, to date, studies about the frequency of autoimmunity in patients with PID have been conducted in groups including adult and pediatric patients. In a small number of pediatric studies, the frequency of autoimmunity was reported to be lower in patients with PID. Lam et al.20 revealed that among the 117 children with PID, autoimmune disorders were found in 6%. Patıroglu et al.21 reported that different autoimmune diseases were detected in 25 patients (4.6%) of the 538 patients with primary immunodeficiency aged 4-27 years old. In a two-center North American cohort with 287 adults and 115 children diagnosed with PID, autoimmunity was reported to be 27.5% in the adult group and 8.7% in the pediatric group.22 Fischer et al.9 reported that immune manifestation of dysregulation, such as autoimmune cytopenia, enteropathy and skin disease, had occurred in 10–30% of children below 18 years of age depending on the underlying immunodeficiency (innate, B cell or T cell defects) and in 40% of patients by age 50 years. In our study, 83 of 822 pediatric patients who were diagnosed with PID had autoimmune/inflammatory manifestations (10.1%).
Autoimmune/inflammatory manifestations may occur in different frequencies according to the PID classifications.23 In the study by Fischer et al., all groups of patients classified as having B cell, T cell and innate immune system deficiencies were found to be at risk of autoimmune and inflammatory manifestations.9 CVID and combined immunodeficiencies were associated with the highest risk. Blazina et al. reported that autoimmune diseases were most prevalent in PIDs classified as diseases of immune dysregulation with 100%, followed by 47% in patients with chronic granulomatous disease and 38% in patients with predominantly antibody deficiencies.19 Defects of phagocytes (56%), combined immune deficiencies (53%) and diseases of immune dysregulation (52%) were the most common diagnostic groups that accompanied autoimmunity in our evaluation. According to Azizi et al.’s study, 26.5% of 471 patients diagnosed with antibody deficiency had autoimmunity.5 In the same study, CVID was the most common primary antibody deficiency that occurred with autoimmunity, with a ratio of 32%. The autoimmunity rate in agammaglobulinemia was 12.7%.24 Coexistence of CVID and autoimmunity was reported at 20-25% in other studies.25–27 Autoimmune cytopenia, pernicious anemia, autoimmune thyroiditis, rheumatoid arthritis and vitiligo were the most common manifestations reported in CVID patients.28–29 In our study, 669 of 822 patients with a diagnosis of PID (81.4%) had primary antibody deficiencies. Coexistences of autoimmunity with CVID and agammaglobulinemia were respectively 21% and 10%, similar to recent studies. When the whole antibody deficiency group was evaluated, it was the group with the lowest rate of autoimmunity and inflammatory symptoms with a rate of 4.3%. It was considered that this result was related to the high ratio of antibody deficiency subgroups such as unclassified antibody deficiency. Patients with hypogammaglobulinemia who do not fulfill all the classical diagnostic criteria for common variable immunodeficiency are categorized as unclassified antibody deficiency.6 Data regarding the clinical presentation, prognosis and treatment of unclassified antibody deficiency patients are limited and the normalization of the immunoglobulin levels with the maturation may be delayed in some of them. On the other hand, it is important to monitor those patients for the probable development of PIDs such as CVID. In the study by Driessen et al.30 comparing CVID patients with unclassified antibody deficiency, it was shown that both groups suffer to a similar extent from infections, while non-infectious complications such as autoimmunity, hepatosplenomegaly or granulomatous disease were only found in CVID patients.
Initial manifestations of autoimmunity/inflammation may be prior to the diagnosis of PID. According to Blazina et al., autoimmune disease was the presenting feature in 42/52 (81%) of all patients with PID who developed autoimmunity.19 Azizi et al. reported that autoimmune manifestations occurred in 46.2% of patients before PID diagnosis, in 17.6% of patients at the time of diagnosis, and in 36.3% of patients in follow-up.24 In a study that included 224 adult and children patients with CVID, registered in the Italian Primary Immunodeficiency Network, the diagnosis of immunodeficiency which was made subsequent to finding autoimmunity in 17.4% and in 2.3% of patients, autoimmune diseases were the only clinical manifestation.25 Two other studies revealed that 54% and 62% of CVID patients had the first episode of ITP or AHA before being diagnosed with immunodeficiency.31–32 In our study, 80.7% of patients (n=67) applied with autoimmune/inflammatory findings or had autoimmunity history before. This occurred in 19.3% (n=16) of patients during follow-up. Early onset or multiple autoimmune/inflammatory manifestations may be the first finding of PIDs despite not having any severe infections. These results reveal the importance of these findings as a warning sign for PIDs.
The type of autoimmunity varies according to the spectrum of immune deficiencies. Data from the US Immunodeficiency Network (USIDNET) have revealed that 1–11% of patients with PIDs have one or more of three autoimmune manifestations: autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, or inflammatory bowel disease.2 A study based on the French CEREDIH PID registry revealed that autoimmune cytopenia was the most common autoimmune disease (31.4%) followed by 24.4% gastrointestinal system findings, 14.1% dermatologic findings, 12.8% rheumatologic findings, 8.1% endocrinologic findings and 3.5% respiratory system findings.23
Autoimmune thyroiditis is the most common autoimmune disorder in the general population and its incidence rate is even higher in certain types of PIDs, including APECED, CVID and IPEX syndrome.5 Autoimmune cytopenia like ITP, autoimmune neutropenia, autoimmune hemolytic anemia and Evans syndrome are common in PID patients and may be the first sign of immune dysregulation, preceding the classical presentation of PID with recurrent infections. A wide spectrum of additional systemic and organ-specific autoimmune diseases has been documented in PID, including diabetes mellitus, non-infectious gastrointestinal diseases (pernicious anemia, celiac disease, autoimmune enteropathy, inflammatory bowel disease), rheumatologic diseases (juvenile rheumatoid arthritis, systemic lupus erythematosus, antiphospholipid syndrome, vasculitis), uveitis, multiple sclerosis, lichen planus, vitiligo, and psoriasis.4,33,34 In our study, autoimmune thyroiditis was the most common involvement (25.3%). This high prevalence may be due to the high level of awareness on this subject and the fact that it is examined during diagnosis and follow-up. Immune thrombocytopenic purpura was the second most common autoimmune manifestation (21.69%) followed by autoimmune hemolytic anemia (19.28%) and neutropenia (12.05%). It is considered that the difference between results may arise from the difference of age and diagnostic groups.
More than one autoimmune/inflammatory manifestation can occur in a patient diagnosed with PID. Fischer et al. reported that 68.1% of patients with PID had only one autoimmune/inflammatory finding.9 In a study by Azizi et al., 69.6% of patients with primary antibody deficiency had one autoimmune manifestation.24 Similarly, 50 patients (60.2%) in our study had one autoimmune/inflammatory manifestation. There was no difference between diagnostic groups according to the number of organs involved. There were multiple autoimmune/inflammatory manifestations in patients with affected gastrointestinal or endocrinological system.
Lymphoproliferation can occur in patients with PID. In one study of 224 CVID patients, prevalence of splenomegaly was noted in 26.4% and it was usually associated with liver enlargement and/or abdominal lymphadenopathy.25 In a study by Azizi et al., patients who had primary antibody deficiency and autoimmunity had lymphadenopathy in 24%, splenomegaly in 37.6% and hepatomegaly in 28% of the cases.24 In our study, lymphoproliferative findings were determined respectively as hepatosplenomegaly (21.7%), lymphadenopathy (12%) and splenomegaly (9.6%) in PID patients with autoimmune/inflammatory manifestations. Phagocytic system defects were the diagnostic group which accompanies lymphoproliferation with the highest rate. The mean age of onset of autoimmune/inflammatory manifestations was statistically higher in patients who had lymphoproliferation coexistent with PID. It was considered that this situation was related with diagnostic groups. It was determined that the presence of lymphoproliferation did not make a difference in terms of prognosis.
A variety of autoantibodies are highly specific for certain autoimmune diseases, but it should be kept in mind that the levels of these autoantibodies in the serum of PID patients such as CVID are often low or negative.5 Barış et al. revealed that direct coombs test was positive in 30.0% of patients; ANA was positive in 16.7% of patients; and thyroid autoantibodies were positive in 13.3% of patients. Autoantibodies were negative in 33.3% of patients.35 Similarly, autoantibody positivity was not determined in 36.1% of patients in our study.
If autoimmune/inflammatory manifestations occur in PIDs, treatment strategies should be targeted to the type of autoimmunity and the pathophysiology underlying the immune deficiency. Balancing immunosuppressive therapy in patients with susceptibility to infections is very important. However, inappropriate treatment of autoimmunity/inflammation increases uncontrolled inflammation and the risk of tissue damage.
In addition to high-dose immunoglobulin therapy, steroids are still accepted as the first line therapy for most autoimmune diseases. Cell growth inhibitors (i.e. azathioprine, cyclosporine, mycophenolate mofetil, cyclophosphamide), and cell depleting monoclonal antibodies (i.e. rituximab) could be used in selected patients as second-line options. Sirolimus for refractory autoimmune cytopenia, anti-TNF for inflammatory bowel disease, CTLA4-Ig therapy for CTLA4 or LRBA deficiency have performed successfully. HSCT or gene therapy may be curative depending on the type of PID.34,36 In our patients, treatment options suitable for PID and the autoimmunity type, especially immunoglobulin and steroid treatment, were used. Twelve patients underwent hematopoietic stem cell transplantation. Autoimmune/inflammatory manifestations in patients with PID are important for prognosis. Baris et al. determined that the survival rate was 70% (n=7) in ten patients in whom HSCT was performed.35 The death rate was determined at 20% in all patients who had autoimmune manifestations. According to Ficsher et al., the overall survival time in patients with PIDs with autoimmune/inflammatory manifestations was found to be significantly shorter than in those without these manifestations.9 The presence of autoimmunity/inflammation before HSCT was associated with a poor survival rate. In our study, the survival rate was determined at 84.3%. The death etiologies of patients were sepsis (46.2%), right cardiac failure (7.7%), development of hemophagocytic lymphohistiocytosis (7.7%), lymphoma (7.7%), liver transplantation complications (7.7%) and post HSCT complications (23%). The survival rate of 12 patients in whom HSCT was performed was 75%. There was no significant difference between survivors and non-survivors in terms of gender, autoimmunity age, age of diagnosis, presence of single or multiple autoimmune manifestations, presence of lymphoproliferation, diagnosis, and organ involvement. The consanguinity rate was significantly higher in parents of the non-survivors.
Although our study was limited by its retrospective nature, it contributes to the literature since it includes a large patient group that includes all types of PIDs in the pediatric age group.
ConclusionEarly diagnosis and treatment of autoimmune and inflammatory diseases in PID pediatric patients are important for increasing quality of life and preventing complications. In our study, the frequency of autoimmunity has been evaluated in a large pediatric patient group consisting of all types of PID patients. As mentioned in our study, in many patients with PID who develop autoimmune/inflammatory manifestations, the findings are observed before or during the diagnosis. For this reason, pediatric patients with autoimmune findings should be evaluated for possible underlying PIDs and patients with known PID diagnosis should also be evaluated for autoimmune and inflammatory disorders in follow up.
Funding informationThis research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Author contributionsMYK, SO, OA, NG collected and analyzed the data; FG, NG conceived and designed the study; MYK wrote the paper; FG supervised the study. All authors approved the final manuscript as submitted.
Conflict of interestThe authors have no conflict of interest to declare.
The authors acknowledge the physicians and patients associated with this study.
