


Common variable immunodeficiency (CVID) is one of the most prevalent forms of primary immunodeficiency characterized by hypogammaglobinemia. Its heterogeneous clinical features include recurrent respiratory tract infections and other complications such as gastrointestinal, autoimmunity, and lymphoproliferative disorders. The aim of this article is to evaluate the general characteristics of CVID patients.
Materials and methodsClinical and immunological features of 44 CVID patients were evaluated retrospectively with long-term follow-up. Patients who participated in the study were diagnosed according to the criteria of the European Society for Immunodeficiency Diseases (ESID).
ResultsThe median age at onset of symptoms was 2.75 years (range 6 months to 17 years), and the median age at diagnosis was 7.75 years (range 4–20 years). The average delay in diagnosis was 4.6 years (range 1–14 years). Positive family history was 18.2%. Before treatment, patients’ median total serum IgG was 271.5mg/dL, median IgA was 7.5mg/dL, and median IgM was 21mg/dL. Infections were the most common clinical manifestation, and 63.6% of patients presented with sinopulmonary infection as the first manifestation. Bronchiectasis developed in 23 CVID subjects, while bronchiectasis was detected prior to CVID diagnosis in eight patients. All patients received immunoglobulin replacement therapy, and one patient died because of granulomatous lymphocytic interstitial lung disease (GLILD).
ConclusionsCVID is a heterogeneous group of immunologic disorders with unknown etiology. There are significant differences in the clinical presentation and prevalence of CVID-related complications among countries. Local guidelines for diagnosis and clinical follow-up are needed.
Common variable immunodeficiency (CVID) is the most common symptomatic primary immunodeficiency disease, characterized by hypogammaglobinemia, recurrent bacterial infections, and chronic complications.1 There are no exact data available, but the estimated prevalence of CVID is between 1:25,000 and 1:50,000, affecting men and women equally.1,2 Although several genes are thought to cause this disease the exact genetic cause remains unknown, and most cases of CVID are sporadic.2,3 The European Society for Immunodeficiencies (ESID) and the Pan-American Group for Immune Deficiency (PAGID) published the diagnostic criteria in 1999. They defined CVID as a patient presenting with low IgG, IgA, and/or IgM levels, onset of immunodeficiency at more than 2 years of age, and impaired antibody response, with the exclusion of other causes of hypogammaglobinemia such as drugs, infectious diseases, or malignancy.4 The International Consensus on Common Variable Immunodeficiency Disorders (ICON) offered new criteria for the diagnosis of CVID in 2015,3 and ESID recently revised its criteria.5 CVID can be seen at any age from early childhood, but it is considered a diagnosis of exclusion and is difficult to distinguish from other immunodeficiencies; therefore, diagnosis should be delayed until the patient reaches at least 4 years of age.1,3
The clinical and immunological spectrum of CVID displays great diversity. Although patients usually present with recurrent respiratory tract infections, many clinical problems including chronic lung disease, gastrointestinal disease, autoimmunity, lymphoproliferative disorders, and malignancy are commonly seen, which cause frequent hospitalization in this patient group.3,6,7 Early diagnosis and treatment are important. The general treatment is immunoglobulin (Ig) replacement therapy, which reduces serious bacterial infections. Along with Ig therapy, any infections that arise must be treated aggressively with antibiotics.8 The clinician must watch carefully for new symptoms and should monitor the patient regularly for complications such as autoimmunity, malignancy, or gastrointestinal problems.8
The aim of the study was to determine the clinical and immunological features of Turkish patients with CVID referred to Istanbul Cerrahpasa Medical School Pediatric Immunology Center over a period of 20 years.
Material and methodsWe retrospectively analyzed the clinical and laboratory findings of 44 patients with CVID disorder who received follow-up in our clinic, Istanbul Cerrahpasa Medical School Pediatric Immunology Division, between 1996 and 2017. The diagnosis of CVID was based on the ESID criteria – that is, they were male or female patients older than 2 years and with decreased levels of at least two serum immunoglobulins (>2 standard deviations below the mean for age) and impaired antibody response, with the exclusion of defined causes of hypogammaglobinemia such as malignancies, medications, protein loss, or bone marrow failure.4 Patients with immunoglobulin deficiency for secondary reasons were excluded from the study. Moreover, other immunodeficiency conditions such as X-linked agammaglobulinemia, X-linked lymphoproliferative disease or combined immunodeficiency were excluded from the study. In our study we used an age of 4 years instead of two, according to the revised ESID diagnostic criteria.5 Patients whose complaints started before the age of four were followed, and if their clinical and laboratory findings upon reaching 4 years of age indicated CVID, they were included in the study.
Each patient's demographic information, family history, clinical manifestations, laboratory investigations (immunological, biochemical, and hematological), and age of diagnosis were recorded. Accompanying co-morbidities, hospitalization, and treatment protocol during the clinical follow-up were recorded from patient files and electronic recording systems.
Blood samples were obtained and placed in ethylenediaminetetraacetic acid (EDTA) tubes for complete blood count analysis (white blood cell count, lymphocyte count, and neutrophil count) and examined using a Beckman Coulter LH780 device. The numbers of CD3+ T, CD4+ T, CD8+ T, and CD19+ B lymphocytes and CD16/56+ NK cells were determined with a Beckton Dickinson FACS Calibur flow cytometer device, using monoclonal antibodies. IgG, A, and M levels were measured using nephelometry with a ROCHE COBAS 702 device, and IgE concentrations were measured using the immunoCAP method. The immunoglobulin and lymphocyte subset levels were determined by comparison with our local age-related normal ranges.9,10 All patients’ immunoglobulin and lymphocyte subset levels were tested before the first Ig replacement therapy. The use of all other diagnostic tests such as chest X-ray, computer tomography of chest, sputum cultures, and hormone level tests was evaluated according to each patient's clinical course as medically required. Expectorated sputum was obtained from patients who had lower respiratory tract infection. The diagnosis of chronic lung disease was made by radiological findings using high-resonance computer tomography.
Follow-up time was measured from the time of diagnosis to March 2017. If a patient's family member displayed symptoms suggesting immunodeficiency that individual was also examined. Approval for the study was obtained from the local ethics committee of Istanbul Cerrahpasa University, Faculty of Medicine (February 2018 – Decision number 1791). Written informed consent was obtained from all participants.
The SPSS program (version 20.0, IBM Company, SPSS Inc.) and Minitab (Version 17.0 software) were used for statistical analysis. p values<0.05 were considered significant. Non-parametric data are expressed using median and range.
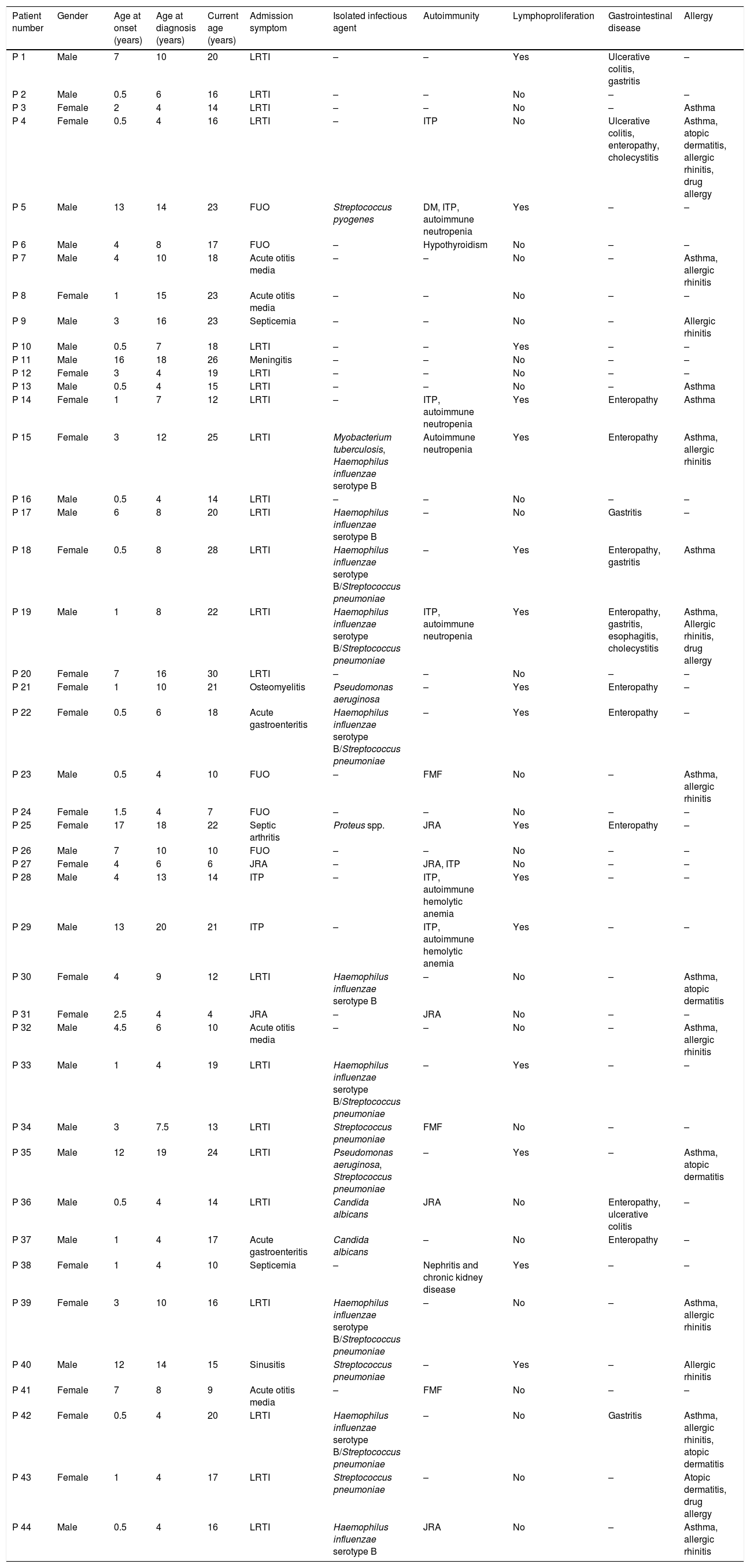
ResultsDemographic features of the patientsThe data of 44 patients diagnosed with CVID were analyzed; 24 (54.5%) were male and 20 (45.5%) were female. The average age at the time of the study was 17 years (range 4–30 years). The median age at onset of symptoms was 2.75 years (range 6 months to 17 years) and the median age at diagnosis was 7.75 years (range 4–20 years). The average delay in diagnosis was 4.6 years (range 1–14 years) and the mean follow-up time was 8.8 years (range 0–20 years). No differences were found between male and female patients in median age at onset of symptoms and at diagnosis (p>0.05). The rate of consanguineous marriage was 40.9% and positive family history was 18.2%. There were two different families in our study who had more than one sibling with immunodeficiency and unaffected parents. The general characteristics of our cohort are given in Table 1.
General characteristics of the patients.
| Patient number | Gender | Age at onset (years) | Age at diagnosis (years) | Current age (years) | Admission symptom | Isolated infectious agent | Autoimmunity | Lymphoproliferation | Gastrointestinal disease | Allergy |
|---|---|---|---|---|---|---|---|---|---|---|
| P 1 | Male | 7 | 10 | 20 | LRTI | – | – | Yes | Ulcerative colitis, gastritis | – |
| P 2 | Male | 0.5 | 6 | 16 | LRTI | – | – | No | – | – |
| P 3 | Female | 2 | 4 | 14 | LRTI | – | – | No | – | Asthma |
| P 4 | Female | 0.5 | 4 | 16 | LRTI | – | ITP | No | Ulcerative colitis, enteropathy, cholecystitis | Asthma, atopic dermatitis, allergic rhinitis, drug allergy |
| P 5 | Male | 13 | 14 | 23 | FUO | Streptococcus pyogenes | DM, ITP, autoimmune neutropenia | Yes | – | – |
| P 6 | Male | 4 | 8 | 17 | FUO | – | Hypothyroidism | No | – | – |
| P 7 | Male | 4 | 10 | 18 | Acute otitis media | – | – | No | – | Asthma, allergic rhinitis |
| P 8 | Female | 1 | 15 | 23 | Acute otitis media | – | – | No | – | – |
| P 9 | Male | 3 | 16 | 23 | Septicemia | – | – | No | – | Allergic rhinitis |
| P 10 | Male | 0.5 | 7 | 18 | LRTI | – | – | Yes | – | – |
| P 11 | Male | 16 | 18 | 26 | Meningitis | – | – | No | – | – |
| P 12 | Female | 3 | 4 | 19 | LRTI | – | – | No | – | – |
| P 13 | Male | 0.5 | 4 | 15 | LRTI | – | – | No | – | Asthma |
| P 14 | Female | 1 | 7 | 12 | LRTI | – | ITP, autoimmune neutropenia | Yes | Enteropathy | Asthma |
| P 15 | Female | 3 | 12 | 25 | LRTI | Myobacterium tuberculosis, Haemophilus influenzae serotype B | Autoimmune neutropenia | Yes | Enteropathy | Asthma, allergic rhinitis |
| P 16 | Male | 0.5 | 4 | 14 | LRTI | – | – | No | – | – |
| P 17 | Male | 6 | 8 | 20 | LRTI | Haemophilus influenzae serotype B | – | No | Gastritis | – |
| P 18 | Female | 0.5 | 8 | 28 | LRTI | Haemophilus influenzae serotype B/Streptococcus pneumoniae | – | Yes | Enteropathy, gastritis | Asthma |
| P 19 | Male | 1 | 8 | 22 | LRTI | Haemophilus influenzae serotype B/Streptococcus pneumoniae | ITP, autoimmune neutropenia | Yes | Enteropathy, gastritis, esophagitis, cholecystitis | Asthma, Allergic rhinitis, drug allergy |
| P 20 | Female | 7 | 16 | 30 | LRTI | – | – | No | – | – |
| P 21 | Female | 1 | 10 | 21 | Osteomyelitis | Pseudomonas aeruginosa | – | Yes | Enteropathy | – |
| P 22 | Female | 0.5 | 6 | 18 | Acute gastroenteritis | Haemophilus influenzae serotype B/Streptococcus pneumoniae | – | Yes | Enteropathy | – |
| P 23 | Male | 0.5 | 4 | 10 | FUO | – | FMF | No | – | Asthma, allergic rhinitis |
| P 24 | Female | 1.5 | 4 | 7 | FUO | – | – | No | – | – |
| P 25 | Female | 17 | 18 | 22 | Septic arthritis | Proteus spp. | JRA | Yes | Enteropathy | – |
| P 26 | Male | 7 | 10 | 10 | FUO | – | – | No | – | – |
| P 27 | Female | 4 | 6 | 6 | JRA | – | JRA, ITP | No | – | – |
| P 28 | Male | 4 | 13 | 14 | ITP | – | ITP, autoimmune hemolytic anemia | Yes | – | – |
| P 29 | Male | 13 | 20 | 21 | ITP | – | ITP, autoimmune hemolytic anemia | Yes | – | – |
| P 30 | Female | 4 | 9 | 12 | LRTI | Haemophilus influenzae serotype B | – | No | – | Asthma, atopic dermatitis |
| P 31 | Female | 2.5 | 4 | 4 | JRA | – | JRA | No | – | – |
| P 32 | Male | 4.5 | 6 | 10 | Acute otitis media | – | – | No | – | Asthma, allergic rhinitis |
| P 33 | Male | 1 | 4 | 19 | LRTI | Haemophilus influenzae serotype B/Streptococcus pneumoniae | – | Yes | – | – |
| P 34 | Male | 3 | 7.5 | 13 | LRTI | Streptococcus pneumoniae | FMF | No | – | – |
| P 35 | Male | 12 | 19 | 24 | LRTI | Pseudomonas aeruginosa, Streptococcus pneumoniae | – | Yes | – | Asthma, atopic dermatitis |
| P 36 | Male | 0.5 | 4 | 14 | LRTI | Candida albicans | JRA | No | Enteropathy, ulcerative colitis | – |
| P 37 | Male | 1 | 4 | 17 | Acute gastroenteritis | Candida albicans | – | No | Enteropathy | – |
| P 38 | Female | 1 | 4 | 10 | Septicemia | – | Nephritis and chronic kidney disease | Yes | – | – |
| P 39 | Female | 3 | 10 | 16 | LRTI | Haemophilus influenzae serotype B/Streptococcus pneumoniae | – | No | – | Asthma, allergic rhinitis |
| P 40 | Male | 12 | 14 | 15 | Sinusitis | Streptococcus pneumoniae | – | Yes | – | Allergic rhinitis |
| P 41 | Female | 7 | 8 | 9 | Acute otitis media | – | FMF | No | – | – |
| P 42 | Female | 0.5 | 4 | 20 | LRTI | Haemophilus influenzae serotype B/Streptococcus pneumoniae | – | No | Gastritis | Asthma, allergic rhinitis, atopic dermatitis |
| P 43 | Female | 1 | 4 | 17 | LRTI | Streptococcus pneumoniae | – | No | – | Atopic dermatitis, drug allergy |
| P 44 | Male | 0.5 | 4 | 16 | LRTI | Haemophilus influenzae serotype B | JRA | No | – | Asthma, allergic rhinitis |
DM: diabetes mellitus, FMF: familial Mediterranean fever, FUO: fever of unknown origin, ITP: idiopathic thrombocytopenic purpura, JRA: juvenile rheumatoid arthritis, LRTI: lower respiratory tract infection.
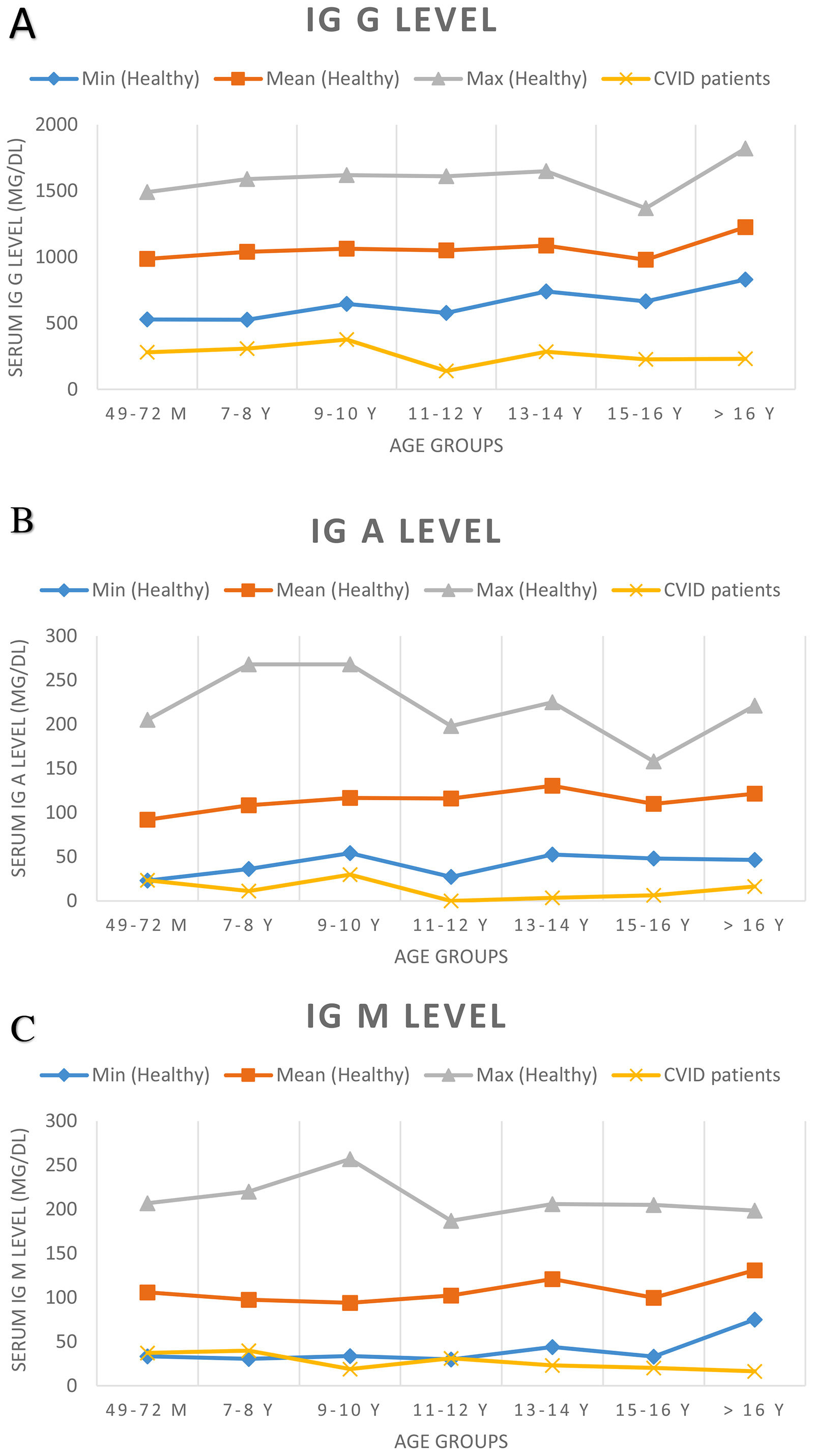
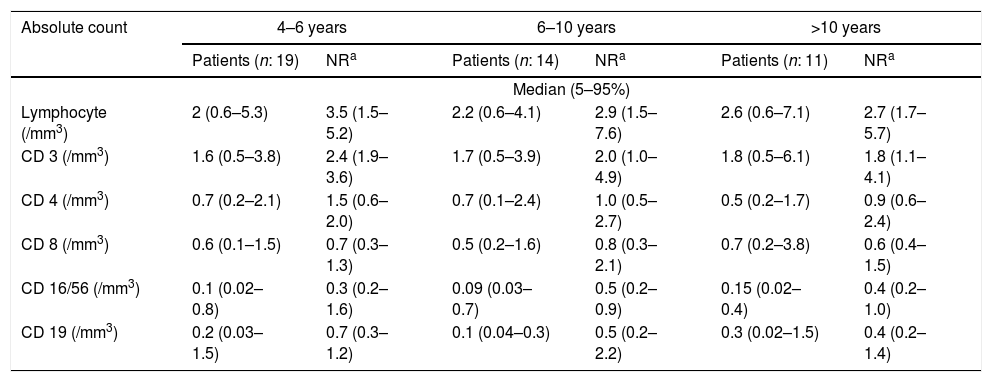
At the time of diagnosis, the baseline serum IgG was set two standard deviations below the normal level for age. Before treatment of the patients, the median total serum IgG was found to be 271.5mg/dL (range 7.0–650mg/dL; four patients had IgG levels of more than 500mg/dL), the median IgA was 7.5mg/dL (range 0–125mg/dL), and the median IgM was 21mg/dL (range 2.9–96mg/dL). IgA levels were found to be under 10mg/dL in 52.2% of the patients. Sixteen patients showed an IgA level of zero. The serum IgM level was less than 25mg/dL for 52.2% of patients. Patients’ serum immunoglobulin level and normal level for age are presented in Fig. 1. The median total lymphocyte count was 2050 cells/mm3 (range 600–7100 cells/mm3). The CD4+ T cell counts of 16 patients were below 500 cells/mm3. The median leucocyte counts are given in Table 2.

Serum immunoglobulin levels of patients and age-related serum immunoglobulin level in healthy children. Normal serum immunoglobulin levels were evaluated according to Ref.9
Absolute number of peripheral blood lymphocyte subsets of patients.
| Absolute count | 4–6 years | 6–10 years | >10 years | |||
|---|---|---|---|---|---|---|
| Patients (n: 19) | NRa | Patients (n: 14) | NRa | Patients (n: 11) | NRa | |
| Median (5–95%) | ||||||
| Lymphocyte (/mm3) | 2 (0.6–5.3) | 3.5 (1.5–5.2) | 2.2 (0.6–4.1) | 2.9 (1.5–7.6) | 2.6 (0.6–7.1) | 2.7 (1.7–5.7) |
| CD 3 (/mm3) | 1.6 (0.5–3.8) | 2.4 (1.9–3.6) | 1.7 (0.5–3.9) | 2.0 (1.0–4.9) | 1.8 (0.5–6.1) | 1.8 (1.1–4.1) |
| CD 4 (/mm3) | 0.7 (0.2–2.1) | 1.5 (0.6–2.0) | 0.7 (0.1–2.4) | 1.0 (0.5–2.7) | 0.5 (0.2–1.7) | 0.9 (0.6–2.4) |
| CD 8 (/mm3) | 0.6 (0.1–1.5) | 0.7 (0.3–1.3) | 0.5 (0.2–1.6) | 0.8 (0.3–2.1) | 0.7 (0.2–3.8) | 0.6 (0.4–1.5) |
| CD 16/56 (/mm3) | 0.1 (0.02–0.8) | 0.3 (0.2–1.6) | 0.09 (0.03–0.7) | 0.5 (0.2–0.9) | 0.15 (0.02–0.4) | 0.4 (0.2–1.0) |
| CD 19 (/mm3) | 0.2 (0.03–1.5) | 0.7 (0.3–1.2) | 0.1 (0.04–0.3) | 0.5 (0.2–2.2) | 0.3 (0.02–1.5) | 0.4 (0.2–1.4) |
NR: normal range.
Many different medical conditions can co-occur as part of the CVID clinical spectrum, as discussed below.
InfectionsRecurrent infections were the most common clinical manifestation and the main complaint of patients. Among the 44 subjects with CVID, 28 (63.6%) presented with sinopulmonary infection as the first manifestation of the disease, involving pneumonia in 23 patients (52.2%), otitis media in four patients (9.1%), and sinusitis in one patient (2.3%). Five patients presented with fever of unknown origin. Less frequent events were gastrointestinal infection in two patients, septicemia in two patients, and one case each of septic arthritis, osteomyelitis, and meningitis.
Two patients who were followed up with for JIA and two followed up with for ITP were diagnosed with CVID during routine laboratory examinations. Nine patients (20.5%) had hearing loss associated with recurrent otitis media.
The patients’ need for hospitalization before immunoglobulin treatment was also examined. Thirty-two patients were hospitalized with a diagnosis of lower respiratory tract disease, seven with acute gastroenteritis, two with meningitis, five with pyelonephritis, four with arthritis, and one with osteomyelitis.
The study included 12 patients diagnosed over 10 years of age. When these 12 patients were compared with others, a lower rate of sinopulmonary infection as the first manifestation of the disease (41.7% vs. 71.9%), a higher rate of bronchiectasis (75% vs. 43.7%), and a similar rate of autoimmunity (41.7% vs. 37.5%) were observed. In addition, the delay in diagnosis was 6.9 years, which was higher than the average of all patients.
Bronchiectasis was present in 23 subjects (52.2%), 12 males and 11 females. In eight of them, bronchiectasis was detected prior to CVID diagnosis. There was no significant difference in IgG and IgM levels between CVID patients who developed bronchiectasis or not (p=0.836 and p=0.133, respectively). However, patients who developed bronchiectasis had a lower IgA level at diagnosis; their mean IgA level was 9.8mg/dL, while patients without bronchiectasis had a mean IgA level of 27.4mg/dL (p=0.025).
GLILD was found in six (13.6%) patients, diagnosed using high-resolution computed tomography (HRCT). One patient who suffered from GLILD died due to respiratory failure. Pathogenic bacteria were detected in sputum cultures from 19 patients (43.2%). The predominant organisms were Streptococcus species and Hemophilus influenzae serotype B (HiB). Mycobacterium tuberculosis was positive in a 21-year-old girl with bronchiectasis who had a severe absence of CD4+ T cells (less than 600 cells/mm3) (Table 1).
Stool cultures were positive for Giardia lamblia in two patients at the gastroenteritis clinic.
Gastrointestinal diseaseThirteen (29.5%) patients had gastrointestinal problems and 10 of them suffered from malabsorption without any unexplained gastrointestinal disorder. Upper gastrointestinal tract endoscopy was performed in four patients; it revealed villous atrophy in two patients while villous atrophy and nodular lymphoid hyperplasia were seen in two. In addition, inflammatory bowel disease (ulcerative colitis) was seen in three patients. Two patients had cholecystitis and cholecystectomy was performed.
AutoimmunityAutoimmune diseases developed in 17 patients (38.6%); the most common one was ITP, detected in seven cases (15.9%). Neutropenia was found in four patients (9.1%), and juvenile rheumatoid arthritis was seen in five patients (11.4%). Different autoimmune conditions such as autoimmune hemolytic anemia, hypothyroidism, and diabetes mellitus type 1 were reported in a few cases. One patient had autoimmune hemolytic anemia and ITP followed as Evans syndrome. Another patient had chronic kidney disease due to nephritis (Table 1). There was no difference in the prevalence of autoimmune diseases between female and male patients (p=0.865). Seventeen patients (38.6%) had splenomegaly; in three of them, it was associated with autoimmune cytopenia and they underwent splenectomy.
AllergyAllergies, such as asthma, allergic rhinitis, atopic dermatitis, and drug allergy, were observed in 18 patients (40.9%). Mean of total IgE level was 17 IU/mL (range 2–1188 IU/mL), and mean of total eosinophil count was 100 cells/mm3 (range 100–1000 cells/mm3).
Other conditions associated with CVIDTonsils were present in nine patients (20.5%). Splenomegaly was detected in 17 patients (38.6%), lymphadenopathy was seen in 12 patients (27.3%), and hepatomegaly was observed in nine patients (20.5%). Eleven subjects (25%) had osteoporosis, verified by dual-energy X-ray absorptiometry (DEXA). Anemia due to iron deficiency was found in 13 patients (29.5%). Growth hormone level just evaluated for 13 patients whose height was under the third percentile. Growth hormone deficiency, diagnosed by a pediatric endocrinologist, was seen in three subjects. Epilepsy was seen in two patients, and one patient had congenital heart disease. No malignancy was observed during follow-up.
Treatment and follow upAll patients were given Ig replacement therapy at the time of diagnosis; 37 (84.1%) were treated with intravenous immunoglobulin (IVIG) and seven (15.9%) were treated subcutaneously. The median Ig dose was 500mg/kg (160–800mg/kg). The Ig dose was adjusted according to the clinical status and immunoglobulin level of the patient. The immunoglobulin dose was increased in patients who had frequent or severe infections. Immunoglobulin levels were controlled at regular intervals; the evaluation time was not standard and changed depending on the clinical course of the patient. Liver and renal function test were checked twice in a year. Anaphylactic reaction developed in four patients (9.1%) due to the IVIG treatment. In two of these patients, there was also non-steroidal anti-inflammatory drugs and β-lactam antibiotics allergy. Anaphylactic reaction occurred in patients with active infection. Sixteen patients (36.4%) who suffered from severe recurrent pulmonary infections were treated with daily antibiotic prophylaxis (trimethoprim/sulfamethoxazole). By 2017, one patient with GLILD had died because of respiratory failure. Another patient gave birth to a healthy child.
DiscussionCVID is a type of primary immunodeficiency disease characterized by different immunological defects and clinical manifestations. The estimated prevalence of CVID in Turkey is 1.39/100,00.11 In this study, we reported 44 CVID patients followed in our center for over 20 years.
Several studies from our country mentioned that the age at onset of symptoms varies between 3 and 7 years while the age at the diagnosis varies between 5.5 and 12 years, including pediatric CVID patients.12–14 In our study, the median age at onset of symptoms was 2.75 years while the median age at diagnosis was 7.75 years. The average delay in diagnosis was 4.6 years. In the literature, this period was found to be between four and five years, similar to our study.6,7,15,16 One possible reason for the delay in diagnosis is the variety across the clinical spectrum of CVID. Patients’ initial referral to the clinic is not always because of recurrent infection. Although a delay in diagnosis may occur due to the clinical findings in many patients being non-specific, eight of our patients had bronchiectasis before CVID diagnosis. This suggests that Ig levels should be evaluated in routine practice in patients with recurrent pulmonary infection.
There is no significant difference in the age of onset, age of diagnosis, immunoglobulin level, or leucocyte count between male and female patients. In one study, female patients with CVID tended to have higher IgM and IgG levels compared to males,6 while in a large cohort study no significant difference was found in IgG levels at diagnosis between male and female patients.7
Consanguinity marriage is a risk factor for autosomal recessive primary immunodeficiency disorders. We found a high frequency of consanguineous marriage (40.9%) and a positive family history (18.2%) in our study cohort. There are several studies from Turkey which obtained similar consanguinity marriage rates and positive family history.11,12,14,17 This can also differ according to region; consanguinity marriage rates are usually high in our country.
In most studies, infections – especially of the respiratory tract – are the most frequent presentation of CVID.2,6,7,15,16,18–20 In our study, respiratory tract infection was the first clinical presentation in more than half of the patients (63.6%), and pneumonia was the most common infection type (52.2%). Studies have obtained different results regarding the frequency of pneumonia and chronic lung disease. Quinti et al.18 found that 49% of patients had suffered at least once prior to diagnosis; but the ratio decreased after starting IVIG replacement (13.3%). In addition, patients with no history of pneumonia before diagnosis have pneumonia under IVIG treatment.18 In a European large-cohort study, pneumonia was seen in 32% of patients, while bronchiectasis was found in 23%.
Bronchiectasis is an important and common medical problem leading to severe respiratory problems.20,21 Even with standard IVIG replacement therapy, bronchiectasis can be seen in many CVID patients, especially those who had low levels of serum Ig and B cells at diagnosis.21 In our study, bronchiectasis was observed in 23 of the patients (52.2%), while in eight patients bronchiectasis developed before diagnosis of CVID. Twelve patients who were diagnosed over 10 years of age, had a high bronchiectasis rate (nine of 12 patients; 75%) according to the rest of our cohort. Resnick et al.2 found the occurrence of bronchiectasis to be 11.2%. Studies showed that bronchiectasis developed with repeated pneumonia was found in 15% of patients and the prevalence of chronic lung disease was more than 50% for the adult age group and 30–40% in the younger population.15,18 Rivoisy et al.22 reported a bronchiectasis rate of 58% for CVID patients with parental consanguinity and 29% for patients with unrelated parents. In a Mexican cohort, bronchiectasis was found in 51% of patients and found more often in patients who suffered from pneumonia.19 The high bronchiectasis rate can be explained by the fact that our clinic is a reference center for pediatric immunodeficiency patients and more complicated cases are followed; even while receiving the IVIG treatment sixteen patients (36.4%) suffered from severe recurrent pulmonary infections. Patients who developed bronchiectasis had significantly lower IgA levels. In addition, Erdem et al.14 reported low IgA level in patients with bronchiectasis compared to patients with other pulmonary complications.
Granulomatous lymphocytic interstitial lung disease developed in 5–15% of CVID patients23 and was related to poor prognosis.23,24 The pathogenesis is unclear.23 In four of the six patients diagnosed with GLILD, recurrent lower respiratory tract infection was observed, despite effective IVIG treatment, and one GLILD patient died due to respiratory failure.
The most commonly isolated infectious agents according to the literature are encapsulated bacteria such as Streptococcus spp. and Haemophilus influenzae, which were seen in our patients also.2,16,21 Rare infections can be seen in clinical follow-up.2,6 In two patients, acute gastroenteritis evolved due to Giardia lamblia, and one patient was diagnosed with pulmonary tuberculosis.
Autoimmune disorders were seen in 20–30% of the patients with CVID.6,7,16,18,19,25 The most commonly seen autoimmune disorders are ITP and hemolytic anemia, which were found in 5–10% of CVID patients, according to the literature.25,26 The main therapy model in the literature is corticosteroids and IVIG. Splenectomy is an option for patients with chronicity or who are not responsive to multiple medical treatments.26 Three patients with autoimmune cytopenia underwent splenectomy in our center. Many autoimmune events have been described in the literature, and it should be noted that in some patients, the autoimmune disorder may be the only finding at the time of diagnosis.19 More than half of patients suffered from cytopenia before the diagnosis of CVID.27 Quinti et al.18 described autoimmune disease as the only clinical manifestation at the time of diagnosis in 2.3% of 224 CVID patients, while in 17.4% of patients autoimmune manifestations presented prior to CVID diagnosis. In a Mexican cohort, two out of 43 (2.8%) CVID patients showed autoimmunity as the first manifestation of disease.19 In our study, 17 patients presented with autoimmunity issues, and in four of them this was the only clinical symptom at the time of diagnosis.
Unlike adult patients, a remarkable finding in the pediatric population is growth hormone deficiency. It can be as high as 25% in the CVID population.28 Growth hormone deficiency was seen in three of our patients, and despite growth hormone replacement treatment, their height was below the third percentile.
The standard treatment for CVID is Ig replacement therapy, which is thought to protect against bacterial infections. However, there is no evidence that it protects against the development of malignancy, autoimmune diseases, or granulomatous diseases.2,7,25,29,30 In a study of 50 patients with CVID, the number of patients experiencing pneumonia decreased from 84% to 22% after treatment with Ig.31 Because some patients had follow-up visits at different hospitals before diagnosis, no detailed information about their previous hospitalizations was documented; thus, the effect of Ig therapy on the patients’ hospitalization rate cannot be evaluated significantly. Lower respiratory tract infection was the most commonly seen infection type after IVIG therapy and a higher dose of Ig replacement and a prophylactic antibiotic approach were applied to patients who had frequent infections.
Both subcutaneous and intravenous routes of administering IgG are effective and maintain adequate IgG levels; however, each has its advantages and disadvantages.32,33 The subcutaneous route benefits from a reduced incidence of systemic adverse events and easier administration; moreover, hospitalization days were decreased and patients have a better quality of life.32,33 The subcutaneous route was only used in seven patients because they only agreed to comply with this treatment.
It has been shown that the immune deficiency status can recover spontaneously over time.6 Our patients’ immunoglobulin levels were periodically checked; no specific timeframe has been identified for spontaneous recovery, and the need for immunoglobulin replacement therapy was not eliminated in any of our patients.
CVID patients have an increased risk of neoplasm, especially lymphoma, compared with the normal population.34,35 The potential causes of this situation are genetic background, immune dysregulation, and chronic clinical infection.29,34 None of our patients developed malignancy that can be explained by malignancy findings that are usually seen in older CVID patients,6 but the average age of our patients was 17 years.
Resnick et al.2 reported a mortality rate in CVID patients of 19.6%, while Quinti et al.18 found a rate of 6%. The mean follow-up period was 8.8 years in our study, and mortality was observed in only one patient. Gathman et al. found that older age at onset and older age at diagnosis are both associated with an increased risk of death. Furthermore, a longer diagnostic delay and presence of a lymphoma or solid tumor is associated with reduced patient survival.7 Low mortality rates may be explained by patient age group and treatment modality. Firstly, Ig replacement therapy is used routinely in follow-up with our patients, and infections are treated effectively with broad spectrum antibiotics. Secondly, most of our patients were in the pediatric age group during the study, and no malignancy was observed.
ConclusionsOur study data are generally compatible with those reported in the literature, but there are significant differences in the prevalence of clinical presentation and CVID-related complications among countries and centers. There may be population-specific differences, or a variation in clinical evaluation. Publication of local data is expected to increase awareness and knowledge among physicians in the country and will help avoid delays in diagnosis. Furthermore, there is a need for international guidelines for the diagnosis and clinical follow-up of patients with CVID.
Conflict of interestThe authors have no conflict of interest to declare.
