


Durante la última década se ha conocido el defecto molecular responsable de un número importante de inmunodeficiencias y este hecho tiene gran repercusión a la hora de establecer un diagnóstico seguro de la enfermedad, dar un consejo genético adecuado y lo que es más importante ha hecho posible el planteamiento de nuevas estrategias a la hora de tratar a estos pacientes. Hasta ahora el tratamiento se basaba en el uso de antibióticos, la administración de gammaglobulina cuando se demostraba una hipogammaglobulinemia o una alteración en la formación de anticuerpos y únicamente en los casos de inmunodeficiencia combinada grave bien definidos, se recurría al trasplante de medula ósea. Además, el mejor conocimiento de la base molecular de muchas de las inmunodeficiencias primarias, no sólo facilita el poder llevar a cabo el tratamiento adecuado, sino que también contribuye a instaurarlo lo más precozmente posible y sin lugar a dudas facilitará el desarrollo de protocolos de terapia génica en muchas de estas enfermedades, como de hecho ya está sucediendo. Cuando se conoce el defecto responsable de la inmunodeficiencia en una familia, las técnicas de biología molecular facilitan en gran manera la detección de portadoras y también, cuando la historia clínica así lo sugiera, el diagnóstico prenatal de la enfermedad en el primer trimestre del embarazo. Esto tiene su indiscutible importancia ya que existe amplia experiencia y bien documentada, de que el trasplante de medula ósea tiene muchas más posibilidades de éxito cuanto más precozmente se realiza e incluso puede hacerse intraútero.
Dentro de las IDP monogénicas las más frecuentes son las ligadas al cromosoma X. Son más numerosas las que se transmiten de manera autosómica recesiva, pero al ser necesaria la falta de función de ambos alelos de un gen sobre cromosomas homólogos, la incidencia es más baja. Además las mutaciones en un determinado gen pueden dar lugar a enfermedades con distinta expresividad fenotípica, no encontrándose en las inmunodeficiencias en general correlación entre fenotipo y genotipo y esto puede ser debido bien a la influencia de otros genes o a factores ambientales.
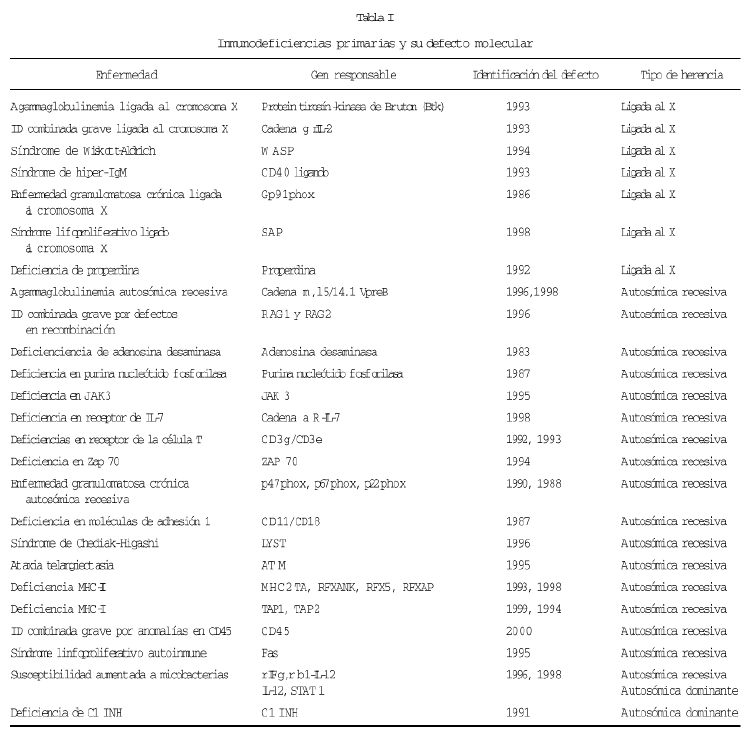
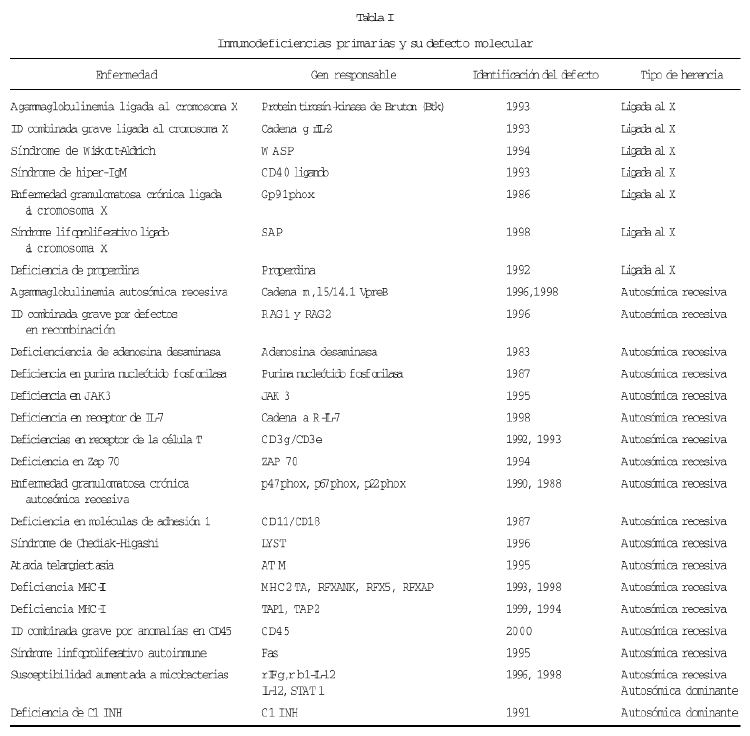
La clasificación de las inmunodeficiencias se ha hecho clásicamente basándose en datos clínicos y alteraciones en el sistema inmune. A estos datos hoy se puede añadir el conocimiento a nivel molecular del defecto que subyace en muchas de estas enfermedades y teniendo esto en cuenta, se están llevando a cabo bases de datos que reúnen gran número de casos con un determinado desorden genético, donde se incluyen la sintomatología, datos de laboratorio, la mutación genética encontrada en cada caso, así como la evolución del paciente con el tratamiento instaurado.
Porque sería demasiado extenso comentar la utilidad del diagnóstico molecular en cada inmunodeficiencia, resumimos en la tabla I las inmunodeficiencias en las cuales, ya se conoce el defecto molecular responsable de las mismas y comentaremos sólo aquellas en las que tenemos experiencia personal sobre su diagnóstico molecular.
AGAMMAGLOBULINEMIA LIGADA AL CROMOSOMA X (ALX)
Es una inmunodeficiencia recesiva ligada al cromosoma X, con un bloqueo en la diferenciación del linfocito B, que se caracteriza por infecciones bacterianas graves y recurrentes así como por enterovirus, ausencia o menos de un 2 % de linfocitos B y agammaglobulinemia (1). El diagnóstico es fácil cuando existen otros varones afectos en la familia, pero en un tercio de los casos no hay historia familiar positiva. Mediante análisis de ligamiento el gen responsable de esta enfermedad fue localizado en el brazo largo del cromosoma X en la región Xq21,3-22 (2), y desde el año 1993 se sabe que las mutaciones en el gen de la tirosin-kinasa citoplásmica denominada tirosin-kinasa de Bruton (Btk), son las responsables de la enfermedad (3, 4). Esta proteína se expresa en linfocitos B y células mielomonocíticas, pero no en linfocitos T.
El gen de la Btk contiene 19 exones que ocupan 37 kb del ADN genómico. El primer exón y 30 pb del segundo constituyen la región no codificante del mismo. La proteína que codifican consta de 659 aminoácidos distribuidos en 5 dominios: el PH (desde el 1 hasta el 137), TH (138-214), SH3 (215-279), SH2 (280-376) y el más largo de todos el dominio kinasa (377-659). La detección de mutaciones en el gen de la Btk se lleva a cabo habitualmente haciendo amplificación de todos los exones y posterior cribaje mediante la técnica de SSCP (5, 6). Si la migración de los fragmentos está alterada sugiere la existencia de una mutación y a continuación secuenciamos el exón amplificado para detectar la mutación responsable. Cuando la SSCP no muestra anomalías y teniendo en cuenta que su sensibilidad es limitada, pasamos a secuenciar todos los exones. Mediante esta tecnología es posible la identificación del 80-90 % de las mutaciones en Btk, pero es preciso tener en cuenta que grandes anomalías como duplicaciones, inserciones, grandes deleciones o inversiones no siempre se detectan con estas técnicas (7). Así tendremos que pensar en la existencia de grandes deleciones cuando nos encontremos con la repetida imposibilidad de amplificar algún exón, y las inserciones y duplicaciones podemos no detectarlas por dicho procedimiento cuando la secuencia del ADN amplificado no esté alterada. De todo esto deducimos que cuando un paciente con fenotipo clínico y analítico compatible con ALX tiene un estudio del gen por SSCP sin anomalías, hay que recurrir a la secuenciación total del gen y también a técnicas de Southern blot. Sólo así sabremos si el 10-20 % restante de pacientes con fenotipo de ALX tienen mutaciones en el gen de la Btk que no son fácilmente detectadas o si lo que ocurre es que se trata de pacientes con fenotipo similar pero debido a anomalías en otros genes. No podemos olvidar la existencia de defectos en la cadena m, en la cadena ligera l 5/14.1 que también dan lugar a agammaglobulinemia congénita con ausencia de células B y fenotipo a veces indistinguible de ALX, pero cuya causa radica en anomalías en otros genes autosómicos (8, 9).
La existencia de ALX con fenotipo atípico menos grave que el clásico, ha hecho que se les haya diagnosticado de manera incorrecta como si de un síndrome variable común de inmunodeficiencia se tratara. De hecho existen en literatura series de SVCID en las que en algunos de los enfermos se ha tenido que modificar su diagnóstico ya que si bien fenotípicamente podía tratarse de ALX no se les consideraba tales al carecer de una historia familiar positiva de la enfermedad (10). Es en estos casos mal diagnosticados de SVCID o déficit de subclases de IgG junto con aquellas agammaglobulinemias autosómicas recesivas (8, 9) que comienzan precozmente con sintomatología y carecen o tienen un número muy bajo de linfocitos B, en los que al poderlos someter ahora al estudio del gen de la Btk se ha podido confirmar o descartar la presencia de anomalías en el mismo.
Las mutaciones encontradas en el gen de la Btk por los diferentes grupos de estudios son muy heterogéneas, generalmente particulares para cada familia y su distribución relacionada con la longitud de cada dominio, por lo que predominan las localizadas en el dominio kinasa (11, 12). Esta misma heterogeneidad afecta al tipo de mutación y mientras algunas dan lugar a un codon stop prematuro que permite predecir la expresión de una proteína truncada, otras lo único que originan es el cambio de un aminoácido por otro (6, 13-15). Para poder confirmar el efecto que estas mutaciones missense tienen sobre la expresión de proteína, es necesario hacer un Western blot en todos estos pacientes.
Nosotros hemos podido encontrar 22 mutaciones diferentes en los pacientes procedentes de este número de familias no relacionadas entre sí y en nuestra experiencia se repite lo descrito en cuanto a tipo de mutación (siete missense, cinco nonsense, tres deleciones, dos deleciones-inserciones, una inserción y cuatro mutaciones puntuales en zonas de splicing) y localización, pues si bien se distribuyen a lo largo de todos los dominios, tienden a agruparse en el dominio kinasa. Historia familiar de ALX hemos encontrado en 11 de los pacientes, aunque todas las madres eran portadoras de la mutación encontrada en su hijo, menos una cuyo hijo tenía una mutación de novo.
Esta tecnología para identificar la anomalía responsable de la enfermedad tiene como inconvenientes ser cara, laboriosa, y cuando no se conoce la mutación en la familia se pueden tardar meses en tener el resultado definitivo. Por este motivo y porque la mayoría de las mutaciones en el gen de la Btk dan lugar a una deficiente expresión de esta proteína, se puede utilizar la citometría de flujo para medirla a nivel intracelular en monocitos (16). Quienes han descrito esta técnica encuentran una ausencia de Btk en los varones afectos, pero en nuestra experiencia este procedimiento no ha sido útil para diagnóstico. Por otra parte el estudio de la proteína mediante Western blot, tiene sus limitaciones ya que algunas mutaciones descritas en el gen no se acompañan de anomalías en la expresión de la proteína (17, 18). De cualquier forma la realización del Western blot permitirá correlacionar tipo de mutación, expresión proteica y fenotipo clínico, todo lo cual llevará a una mejor caracterización de estos pacientes.
Antes de conocerse el gen de la Btk, el estudio de portadoras se hacía determinando la inactivación del cromosoma X. Las mujeres portadoras de inmunodeficiencias ligadas al cromosoma X presentan una inactivación sesgada del mismo en la línea celular afectada por el defecto y todas estas células tendrán como activo el cromosoma X que porta el alelo sano, mientras que en el resto de las células la inactivación es al azar. Este estudio es importante para asegurar el diagnóstico en casos atípicos de ALX o esporádicos, teniendo siempre en cuenta que una inactivación al azar no excluye la posibilidad de una mutación de novo en línea germinal materna (19). Más recientemente el estudio de portadoras se ha intentado hacer por citometría de flujo viendo la expresión intracelular de Btk en monocitos, que mostrará un patrón de tinción bimodal característico tras incubación de los mismos con antisuero monoclonal anti Btk (16). El Western blot sólo confirmará el estado de portadora en las madres de pacientes de ALX que expresen una proteína Btk de tamaño anómalo, en cuyo caso la condición de portadora se confirmará por la presencia de dos bandas de diferente tamaño.
Pero si el estudio molecular es definitivo a la hora de hacer un diagnóstico de ALX, no lo es menos en el diagnóstico de portadoras y es más seguro que la tecnología anteriormente descrita. Mediante SSCP podemos ver un patrón característico de movilidad alterada y tras secuenciación, la presencia de una imagen doble por el solapamiento del alelo sano y el mutado encontrado en la familia.
El diagnóstico prenatal, cuando existan antecedentes familiares de la enfermedad, se basará en la presencia o no de linfocitos B en sangre fetal extraída hacia la 18-20 semana de la gestación o bien buscaremos la mutación en el ADN obtenido con biopsia de corion realizada en la novena semana de gestación o en líquido amniótico unas semanas más tarde.
SINDROME DE HIPER-IgM LIGADO AL X
Está incluida dentro de las inmunodeficiencias combinadas y en primer lugar para su diagnóstico correcto se valorará el cuadro clínico. En los primeros años de vida presentan afectación sinupulmonar y en algunos pacientes el primer síntoma es una anemia aplásica inducida por parvovirus (20) o una neumonía por Pneumocystis carinii. Con frecuencia tienen episodios diarreicos producido por Critosporidium y los pacientes de más edad colangitis por el mismo germen, siendo un dato analítico frecuente la presencia de neutropenia. Hay un fenotipo clínico más benigno en función de la edad de comienzo de los síntomas, frecuencia con la que requieren tratamiento antibiótico, que tengan colangitis, Pneumocystis, neutropenia y su respuesta al tratamiento con gammaglobulina intravenosa.
Desde el punto de vista analítico tiene cifras normales o elevadas en suero de IgM y muy disminuidas las de IgG e IgA. Por citometría de flujo podemos ver cómo sus linfocitos B sólo llevan en superficie IgM e IgD y carecen de células B memoria (IgD-CD27 +) fenotipo muy similar a las células B de cordón umbilical, con capacidad escasa de producir IgG e IgA (21). El porcentaje de linfocitos T es normal y con capacidad para responder a mitógenos pero su respuesta proliferativa a antígenos está disminuida.
Una sintomatología sugestiva y los datos de laboratorio descritos harán sospechar la enfermedad. En el año 1993 grupos de trabajo diferentes (22, 23) demostraron que el síndrome de HIM-X se debe a defectos en el gen que codifica la molécula del CD40L (CD154), glucoproteína de membrana que expresan transitoriamente y no de manera constitutiva, los linfocitos T CD4+ activados y en menor proporción en los T CD8+, mastocitos, basófilos y plaquetas. El CD40 es otra glucoproteína que se expresa en la superficie de las células B, monocitos, células dendríticas y epiteliales y cuya unión con el CD40L es necesaria para que las células productoras de IgM pasen a células productoras de IgG e IgA. La unión CD40-CD40L tiene también un papel mal definido en la función de los linfocitos T.
El diagnóstico por citometría se basa en la expresión del CD40L sobre células T previamente estimuladas y posterior tinción con anticuerpos monoclonales. También puede visualizarse el CD40L por citometría mediante una proteína de fusión o constructo (CD40-Ig), formada por la región extracelular del CD40 humano y la porción Fc de la Ig de ratón. La incapacidad de las células T activadas para unirse con anticuerpos monoclonales anti-CD40L o a la proteína CD40-Ig, será de gran ayuda para el diagnóstico del síndrome de hiper-IgM, ya que en los afectados sus células activadas no expresan el CD40L (4 % de los casos) o lo hacen con menor intensidad que los controles sanos (24, 25).
El gen del CD40L se localiza en el cromosoma X, Xq26.3, Xq27.1 y consta de cinco exones. El primer exón codifica el dominio intracelular, el transmembrana y parte del extracelular, estando el resto codificado por los otros exones. Como en el caso de la ALX el estudio molecular comienza con la amplificación a partir de ADN genómico de los cinco exones y sus zonas intrónicas limítrofes. Cuando las mutaciones se localicen en la zona aceptora o donadora de splicing estudiaremos la repercusión sobre el procesamiento del ARN inmaduro haciendo una RT-PCR. En un paciente con HIM-X, cuyo diagnóstico inicial fue de SVCID, pudimos comprobar tras realizar el estudio molecular que tenía una mutación puntual en el gen del CD40L. En otra familia que hemos podido estudiar con dos hermanos afectos, encontramos una mutación en el intrón 3 que daba lugar a la pérdida del exón 3 a nivel de cADN. Hemos visto por citometría de flujo que los tres expresan el CD40L sobre sus linfocitos T activados, pero en menor intensidad que los controles sanos. Este patrón de tinción es similar al que se encuentra en algunos pacientes con síndrome variable común de inmunodeficiencia (26) en cuyo caso hay que hacer con ellos el diagnóstico diferencial, especialmente si no existen en la familia otros varones afectos de HIM-X. Esta es una de las razones por las que si bien la citometría es muy útil para el diagnóstico de esta forma de HIM-X, será necesario confirmarlo mediante estudios moleculares. Sólo así el consejo genético será adecuado como la instauración del tratamiento definitivo hoy recomendado, que consiste en el trasplante de medula ósea lo más precozmente posible, en los casos en que exista donante idéntico.
No se ha encontrado correlación entre mutaciones específicas y clínica, existiendo incluso una considerable variabilidad intrafamiliar en el fenotipo clínico. De hecho en los hermanos anteriormente comentados, el menor tiene un fenotipo clínico más benigno que el mayor.
Ya que el defecto genético no es letal para las células T, las portadoras tienen inactivación al azar del cromosoma X en sus células T por lo que en citometría la expresión del CD40L dará una tinción bimodal, aunque puede existir un sesgo importante. Por este motivo para el diagnóstico de portadora será definitivo el estudio molecular y así hemos podido llegar a este diagnóstico en la madre y hermana de tres varones fallecidos con síndrome compatible con HIM-X.
Para el diagnóstico prenatal de la enfermedad, cuando se conozca el defecto molecular en la familia, se buscará la mutación en ADN obtenido de vellosidades coriónicas. Debido a la escasa capacidad de las células T del recién nacido para expresar CD40L, la expresión del mismo en la sangre fetal extraída a las 20 semanas, no servirá como técnica aislada para hacer el diagnóstico intraútero.
Existe otro síndrome de hiper-IgM con herencia autosómica recesiva (HIGM2), pero en estos pacientes la secuenciación del gen del CD40L así como la expresión sobre la membrana celular de esta glucoproteína son normales (27). Recientemente se ha descrito que mutaciones en el gen de la activación inducida por citidín desaminasa (AID), son las responsables de este síndrome (28).
El diagnóstico molecular en estas inmunodeficiencias como en todas aquellas cuyo defecto genético hoy es conocido, permitirá hacer un diagnóstico más seguro e implantar en cada caso el tratamiento más adecuado. Además el conocimiento de estos defectos ayudará a conocer mejor la patogenia de las inmunodeficiencias primarias.
AGRADECIMIENTOS
Agradecemos a Priscila Acebes Sanz, Milagros Arribas Escaso, Amparo Barranco Herrero, Josefa Borrega Guillén, Ana Carazo Álvarez, Begoña Castro Rodríguez y Josefina Moruno González la ayuda técnica para el desarrollo de este trabajo.
