


The Hyper-immunoglobulin M syndromes (HIGM) are a heterogeneous group of genetic disorders, which have been rarely reported to be associated with growth hormone deficiency (GHD).
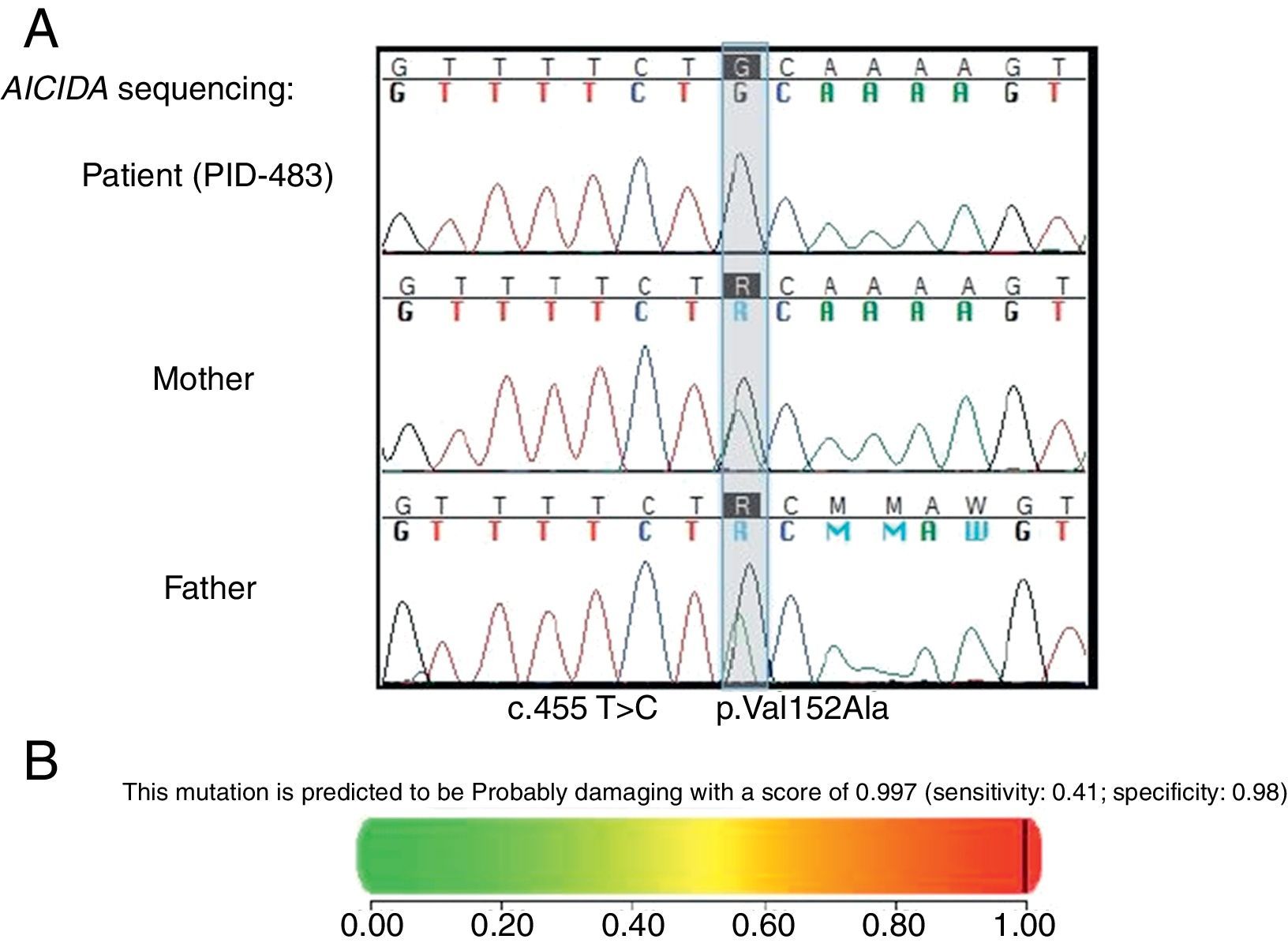
Methods and resultsA nine-year-old girl with recurrent urinary tract infections, diarrhoea, sinopulmonary infections, and failure to thrive since the age of six months had normal CD3+, CD4+, CD8+T lymphocytes, and CD19+B lymphocytes and natural killer (NK) cells, but extremely elevated IgM and significantly decreased IgG and IgA. In view of the patient's short stature, growth hormone evaluation was carried out and growth hormone deficiency established. The patient underwent Ig replacement therapy and received growth hormone therapy in addition to antibiotics and responded well. Furthermore, the patient developed benign cervical lymphadenopathy, as well as elevated erythrocyte sedimentation rate, positive autoantibodies to SSA-Ro, and severely dry eyes, which partially responded to both the punctate occlusion and systemic corticosteroids, at the age of seven years. Sequencing analysis of the exons from activation-induced cytidine deaminase (AICDA) gene revealed that the patient was homozygous for a single T to C transversion at position 455 in exon 4, which replaces a Valine with an Alanine.
ConclusionsTo our knowledge, this is a new AICDA mutation, which has not been reported previously in HIGM. The mutation analysis could improve diagnosis of HIGM patients and also elaborating on the spectrum of AICDA mutations.
Hyper-immunoglobulin M (HIGM) syndromes or immunoglobulin class switch recombination deficiencies (CSR-Ds) are rare primary immunodeficiency (PID) disorders, characterised by normal to high IgM levels and decreased serum levels of IgG, IgA and IgE due to defects of class switching recombination, with or without an impairment of somatic hypermutation (SHM), depending on the molecular defect.1 Following the discovery of the mutations in the genes encoding CD40 ligand (CD40L) to be involved in patients with HIGM in 1993,2 more than 200 gene defects have been identified to take part in CSR-Ds pathogenesis.3,4 These defects could be categorised to different groups based on their way of action, such as the defects in T cell-B cell cooperation, intrinsic B cell defects, as well as DNA repair defects.5 The most renowned gene defects resulting in imperfect T cell-B cell cooperation, comprises those leading to defective CD40L, which is the most frequent condition causing CSR-Ds, with an X-linked inheritance (type 1 HIGM)6; CD40 deficiency with autosomal recessive inheritance (type 3 HIGM)7; impaired nuclear factor-kappa B (NF-κB) activation, resulted from nuclear factor-kappa-B essential modulator (NEMO) gene mutations, with an X-linked inheritance (type 6 HIGM)8; inducible costimulator (ICOS) molecule deficiency with autosomal recessive inheritance9; and an uncharacterised CSR-D with normal in vitro CSR, which is suspected to be caused by the defective generation of T follicular helper cells or their activation or interaction with follicular B cells.5 The other category, intrinsic B cell defects, could be caused by either the mutations in the activation-induced cytidine deaminase (AICDA) gene culminating in the most frequent autosomal recessive CSR-D, the activation-induced cytidine deaminase (AID) deficiency (Type 2 HIGM),10 or a lack of cytoplasmic co-factor of AID.11 The last above-mentioned category, DNA repair defects, could be resulted from autosomal recessive Uracil-N-glycosylase (UNG) deficiency (type 5 HIGM),12 autosomal recessive post-meiotic segregation 2 (PMS2, which is a member of mismatch repair (MMR) complex of proteins) deficiency,13 autosomal recessive ataxia telangiectasia,14 or an unknown DNA repair deficiency.5 The genetic basis of many other cases of HIGM has not been identified thus far (type 4 HIGM).
The humoral immunodeficiency resulted from CSR-Ds leads to an increased susceptibility to bacterial infections, affecting mostly the gastrointestinal and respiratory tracts. Furthermore, marked lymphoid hypertrophy, with the characteristic giant germinal centres, is another prominent clinical feature of AICDA deficiency, which has been reported in 75% of cases and may occur as a consequence of microbial infection.15 Similarly, it has been described in the other forms of CSR defect.16
Although growth hormone deficiency (GHD), resulting in short stature, has been previously reported to be associated with a number of immunodeficiency states, its association with HIGM is very rare, and to the best of our knowledge has only been reported in two patients (Japanese and Iranian) thus far.17,18
Here we report a case of GHD with an autosomal recessive HIGM by phenotype and genotype, with a novel mutation in AICDA that has not been reported formerly.
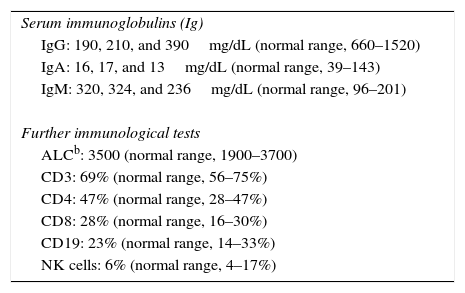
Methods and resultsThe patient was a nine-year-old girl born in 2007 via caesarean section following an uneventful full-term gestation without perinatal problems, and received routine vaccine inoculations without any obvious complications. Her parents were consanguineous. She has no siblings. There was no history of immunodeficiency disease or recurrent infections in the family. The patient remained in good health until the age of six months, when she gradually developed failure to thrive and recurrent urinary tract infections that were treated with antibiotics, but continued until 18 months of age, despite receiving prophylactic antibiotic (Trimethoprim/sulfamethoxazole) for 12 months. During these months, she continued to have recurrent urinary tract infections, experienced repeated episodes of diarrhoea, developed sinopulmonary infections and chronic otitis media, and failed to thrive. Consequently, she was referred to the immunology department at Namazi Hospital in Shiraz, Iran with suspected immunodeficiency at the age of two years. Both the abdominopelvic ultrasound and voiding cystourethrogram failed to reveal any abnormal findings. Laboratory results were as follows: blood routine examinations showed haemoglobin, 12g/dL (within the normal range for age); and normal counts for white blood cells (WBC) (WBC, 9800/mm3; lymphocytes, 36%; neutrophils, 56%) and platelets (270,000/mm3). Urine analysis revealed many bacteria as well as many WBCs, which was subsequently confirmed by the positive urine culture of more than 105 colony forming units of Klebsiella spp. Immunological investigations depicted low serum IgA and IgG levels but increased serum IgM levels in repeated measurements (Table 1). On the basis of the aforementioned findings, a diagnosis of HIGM syndrome was made and immunoglobulin replacement therapy and antibiotics were started. Bilateral T-tubes were also inserted to control the chronic otitis media. Further immunological work-up divulged normal numbers for CD3+ T cells, CD4+, and CD8+ T cells (measured by flow cytometry), CD19+ B cells, and natural killer (NK) cells (Table 1). Genetic studies showed a novel homozygous missense mutation in the coding region of AICDA, which has not been reported previously (Fig. 1).
Laboratory test results.
| Serum immunoglobulins (Ig) |
| IgG: 190, 210, and 390mg/dL (normal range, 660–1520) |
| IgA: 16, 17, and 13mg/dL (normal range, 39–143) |
| IgM: 320, 324, and 236mg/dL (normal range, 96–201) |
| Further immunological tests |
| ALCb: 3500 (normal range, 1900–3700) |
| CD3: 69% (normal range, 56–75%) |
| CD4: 47% (normal range, 28–47%) |
| CD8: 28% (normal range, 16–30%) |
| CD19: 23% (normal range, 14–33%) |
| NK cells: 6% (normal range, 4–17%) |
![(A) Mutation found in AICDA. The coding regions (±10 base pairs) of the AICDA gene (NM_020661) has been PCR amplified and sequenced. A homozygous single nucleotide variant has been identified at position c.455T>C in exon 4, which resulted in a homozygous missense mutation at position p.Val152Ala of the corresponding protein product. (B) PolyPhen-2 report for the pathogenicity of the amino acid substitution p.Val152Ala in AICDA[19].](https://static.elsevier.es/multimedia/03010546/0000004500000001/v1_201612300037/S0301054616301161/v1_201612300037/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNe5umqYt0zKZP4SYniTZ7+SgZrkCgayp3MRA1ovxa2vU7XiyYWZcFWkqgjpVKRJ9poVaj3SBBsOthc7Ur7UDb60RlEk+6ged2Lgjv6jd3S69O3VwWdQF3ga+3WS6/e2bYcTAKc946ivRiAtJiJUj6e27FlqH33lsOav4dL2BiaeRWHdbEaRy7rw6B133SOsoMrDSb4QZrbUZuoxQy0U/+zTbwPjz1s2cywDRIcwgJ7qapQwgmnXL0q2nmgf8Dhj5AXZ7MOUFRMLIO9W1BBMwuUp "(A) Mutation found in AICDA. The coding regions (±10 base pairs) of the AICDA gene (NM_020661) has been PCR amplified and sequenced. A homozygous single nucleotide variant has been identified at position c.455T>C in exon 4, which resulted in a homozygous missense mutation at position p.Val152Ala of the corresponding protein product. (B) PolyPhen-2 report for the pathogenicity of the amino acid substitution p.Val152Ala in AICDA[19].")
(A) Mutation found in AICDA. The coding regions (±10 base pairs) of the AICDA gene (NM_020661) has been PCR amplified and sequenced. A homozygous single nucleotide variant has been identified at position c.455T>C in exon 4, which resulted in a homozygous missense mutation at position p.Val152Ala of the corresponding protein product. (B) PolyPhen-2 report for the pathogenicity of the amino acid substitution p.Val152Ala in AICDA[19].
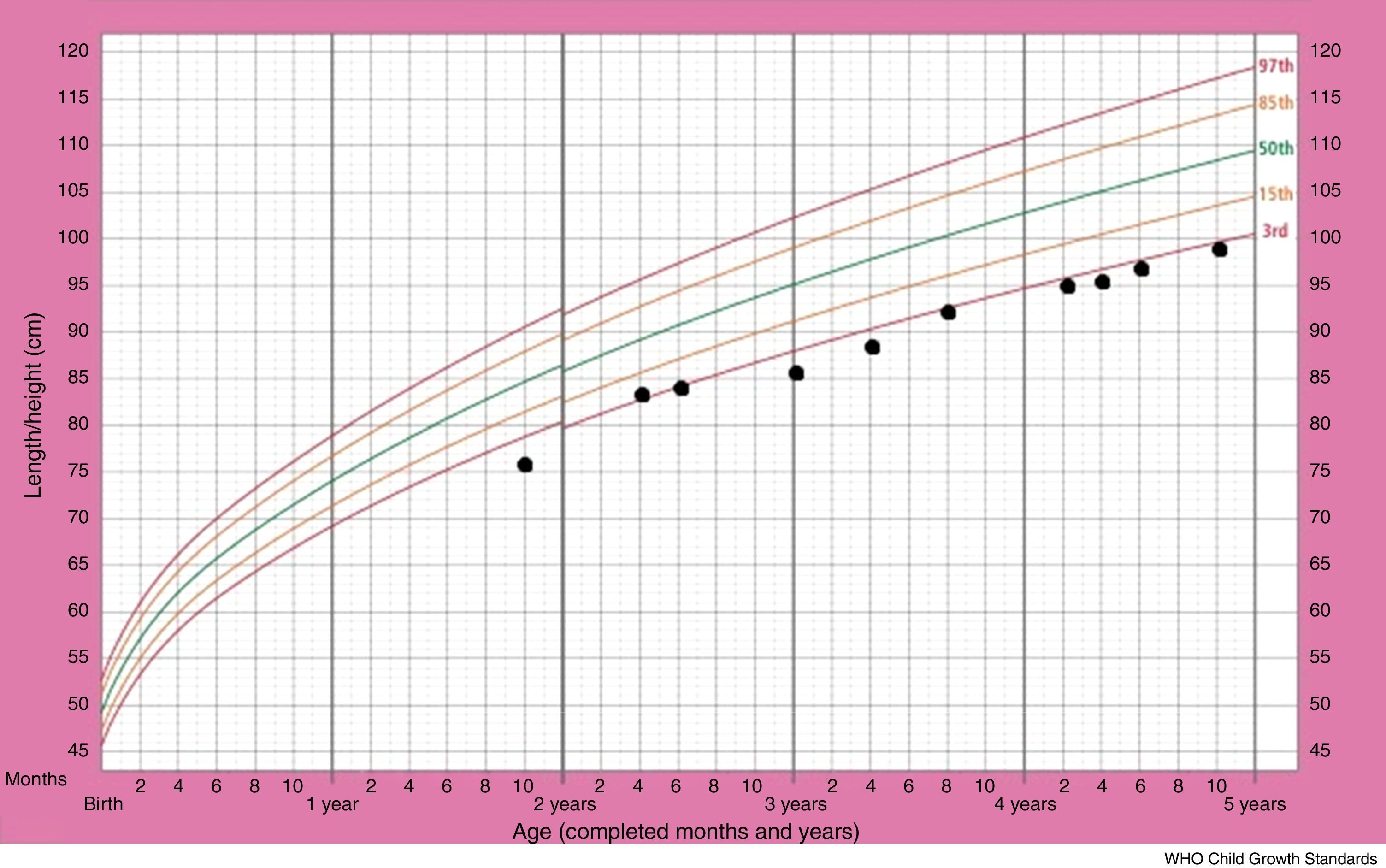
Although the patient improved clinically with treatment, her growth retardation continued. The patient's stature-for-age chart has been depicted in Fig. 2. At the age of three years, the patient's weight was between the 5th and 10th percentile for her age, and her height for age was just below the 3rd percentile, with the growth rate of less than 2cm over a six-month span. The bone age of two years has been estimated from the left wrist and hand X-ray, at the chronological age of three years. The initial laboratory investigations including serum electrolyte concentrations (Na, K, Cl, P), fasting blood sugar, blood urea nitrogen, creatinine, urine analysis as well as certain hormone tests, comprising thyroxine, thyroid stimulating hormone and cortisol, yielded results within normal ranges. The aspartate aminotransferase (AST) level was 29U/L (normal range: 1–46) and the alanine aminotransferase (ALT) level was 15U/L (normal range: 1–49). On the other hand, growth hormone evaluation confirmed growth hormone deficiency. Growth hormone (GH) levels measured using l-dopa and clonidine, were low (GH of less than 10ng/ml in response to the provocative agents) (Table 1). The patient was given growth hormone therapy during four consecutive months, which resulted in a growth rate of 6cm over a year. However, the patient discontinued therapy due to the high cost.

She has also experienced several episodes of large cervical lymphadenopathies since she was seven years old. Biopsy examination demonstrated reactive follicular hyperplasia.
The patient has recently developed severe eye dryness, positive anti-SSA/Ro antibody, and elevated erythrocyte sedimentation rate. Low dose systemic corticosteroid and standard local eye care could not improve the clinical outcome. However, punctate occlusion culminated in partial improvement of eye dryness. Cyclosporine eye drops have been added recently, as well.
DiscussionActivation-induced cytidine deaminase deficiency, the second identified genetic cause of HIGM syndrome, is caused by AICDA gene mutations. Following stimulation through CD40 and cytokines, AID is expressed transiently and selectively in germinal centre B-cells and plays a crucial role in B-cell terminal differentiation through deamination of cytidine into uracil residues, which results in the induction of DNA lesions in both the switch (S) and variable (V) regions of immunoglobulins in the early phase of CSR and SHM. A lack of AID culminates in the defective CSR and SHM, either normal or high titres of IgM, as well as absent or very low levels of IgG, IgA and IgE. Nevertheless, the proportion of memory B cells expressing CD27 is present in normal numbers.10 Defect in both CSR and SHM could be resulted from the mutations scattered throughout the AICDA gene.20 Complete lack of CSR but normal SHM could be a consequence of mutations positioned in the C-terminal domain of the AICDA gene.21 On the other hand, heterozygous nonsense mutations in the C-terminal part of AICDA gene has been reported to cause a variable, autosomal dominant CSR-D.5,15
Our patient presented with severe eye dryness, together with positive anti-SSA/Ro antibody, both of which showed suggestive evidence for autoimmunity. Autoimmune complications, including haemolytic anaemia, thrombocytopenia, hepatitis, and arthritis have been formerly described in about 20% of AICDA deficient HIGM patients.22 The presence of IgM autoantibodies, defective development of regulatory T cells, impaired peripheral B-cell tolerance checkpoint, expansion of autoreactive B lymphocytes as a result of the immature and transitional B cells resistance to apoptosis, activation of autoreactive T cells by presenting self-antigens by B cells (62), as well as the development of ectopic lymphoid tissue in non-lymphoid organs, which itself results from B cell lymphoproliferation, and eventually predisposes to organ specific autoimmunity, have been implicated as the potential mechanisms for the autoimmune process in HIGM syndrome.15,16,23
Despite the fact that drastic growth impairment has been enunciated in HIGM, the association with this entity has been exclusively reported in two cases to date. This phenomenon was first described in a Japanese patient in 1993.17 Ohzeki et al. reported a case of hyper-IgM immunodeficiency concomitant with growth hormone deficiency, and suggested the autosomal dominant mode of transmission.17 Kashef et al. described an Iranian patient in whom the association between growth hormone deficiency and HIGM syndrome had been verified in 2009.18 However, they announced all four known genes responsible for immunoglobulin CSR-Ds to be intact.18 Here, we explain the association in an Iranian patient in whom the autosomal recessive mode of transmission, involving a missense mutation at position c.455T>C in exon 4 in AICDA gene, has been justified. The GH gene is known to be positioned on the long arm of chromosome 17 (q21.0–q22.0).24 It is arduous to describe the genetic association between the AICDA deficient CSR-D, whose gene is located on the chromosome 12, and hereditary GH insufficiency. It could be postulated that the two conditions occur coincidentally. However, it may be speculated that GH deficiency is associated with autoimmunity as a consequence of AICDA deficiency. A probable hypothesis could be the presence of autoreactive pituitary antibodies (APA) against pituitary growth hormone-secreting cells suggesting an autoimmune pituitary involvement in such cases. Nevertheless, the other aforementioned autoimmune processes in AICDA deficient patients could be hypothesised to be partly responsible in the autoimmune pituitary involvement as well. However, the possible underlying mechanism leading to CSR-D, autoimmunity and growth hormone deficiency needs to be elucidated.
In conclusion, we report an unusual mutation in AICDA, in a case of HIGM syndrome, growth hormone deficiency, and autoimmunity. Notwithstanding the fact that we could not completely rule out that these conditions are coincidental events, this case is meaningful because the combination is eminently rare.
Ethical disclosuresConfidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
FundingThis study has no funding source.
Conflict of interestAuthors declare no conflicts of interest.
It should be noted that there is no ethical problem (approved by the research ethics committee of Tehran University of Medical Sciences) or conflict of interest in our research. There was not any honorarium, grant, or other form of payment to authors to produce the manuscript.
