


All allergen products for allergen immunotherapy currently marketed in the European Union are pharmaceutical preparations derived from allergen-containing source materials like pollens, mites and moulds. Especially this natural origin results in particular demands for the regulatory requirements governing allergen products. Furthermore, the development of regulatory requirements is complicated by the so far missing universal link between certain quality parameters, in particular biological potency, on the one hand and clinical efficacy on the other hand. As a consequence, each allergen product for specific immunotherapy has to be assessed individually for its quality, safety and efficacy. At the same time, biological potency of allergen products is most commonly determined using IgE inhibition assays based on human sera relative to product-specific in house references, ruling out full comparability of products from different manufacturers. This review article aims to summarize the current quality requirements for allergen products including the special requirements implemented for control of chemically modified allergen extracts (allergoids).
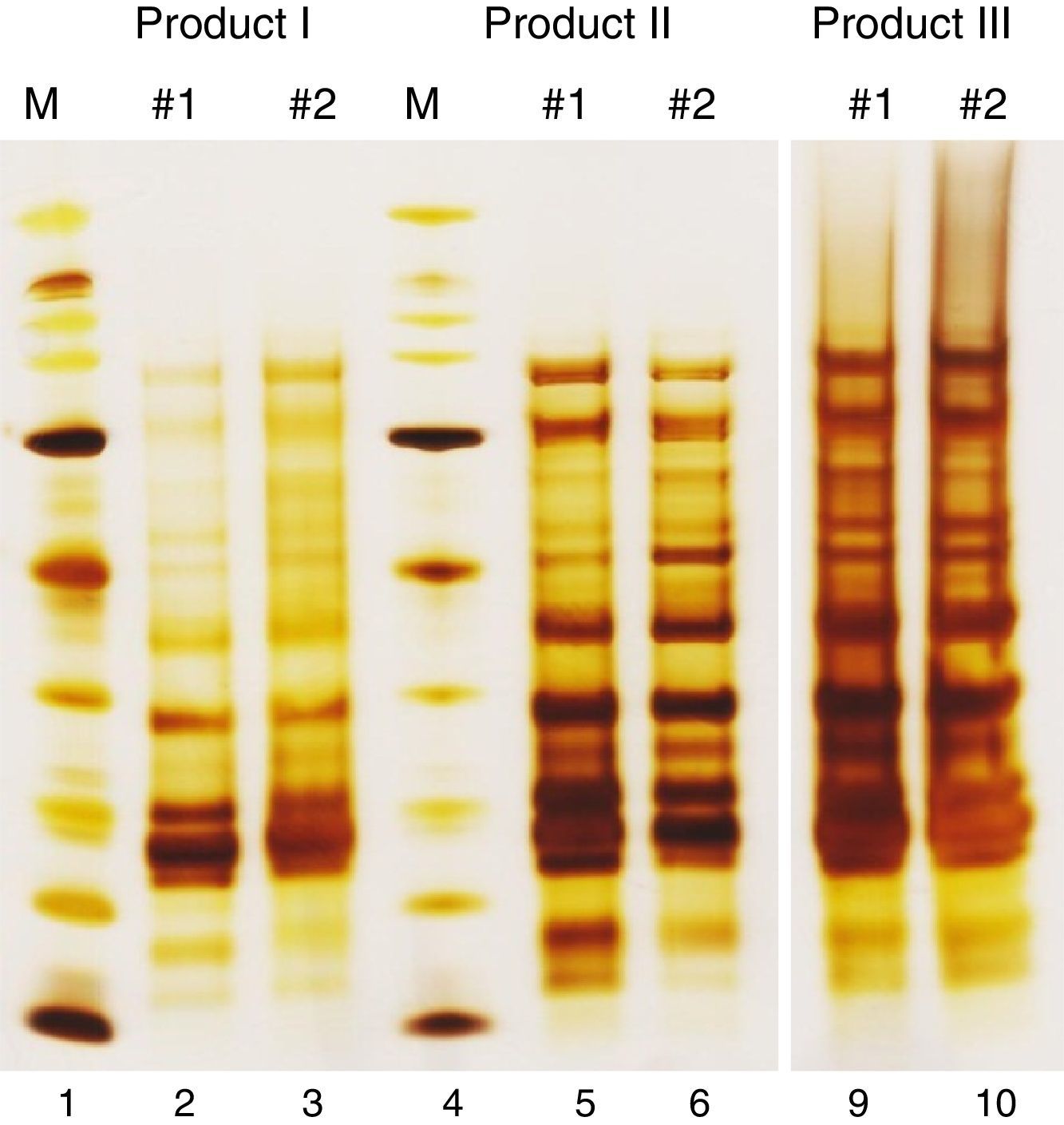
Like all industrially produced medicinal products, products for allergen-specific immunotherapy (AIT) require a marketing authorization for distribution in the European Union (EU). However, several characteristics distinguish AIT products from other medicinal products, necessitating special regulatory requirements.1 Examples for these special characteristics are the wide variety of allergenic substances, the widespread use of chemically modified and/or adsorbed allergen extracts and the bio-variability of the natural source materials. Furthermore, each AIT product has to be regarded as unique, even if products originate from the same source material. For example, there are currently 13 AIT products available on the German market for treatment of birch pollen allergy. Although the active substances in all of these products originate from aqueous extracts prepared from pollen of the tree species Betula verrucosa, each of these products has to be assessed individually for its quality, safety and efficacy. Among other reasons, this is necessary because differences in the manufacturing processes from the natural source materials to the drug products may lead to considerable differences in protein composition (Fig. 1).
 are presented. The molecular weight marker (M) ranges from 6.5kDa to 200kDa. Two empty lanes of the gel (7 and 8) have been cut from the picture to allow for better side-by-side comparison.")
Protein profiles of birch pollen allergen extracts. SDS-PAGE under reducing conditions was performed with native intermediates of three birch pollen allergen products from different manufacturers. Of each product, two batches (#1, #2) are presented. The molecular weight marker (M) ranges from 6.5kDa to 200kDa. Two empty lanes of the gel (7 and 8) have been cut from the picture to allow for better side-by-side comparison.
In addition, the natural origin of the allergen source materials entails the problem of bio-variability in both allergenic as well as non-allergenic components. For example, Codina et al. recently summarized the knowledge on factors reported to influence the composition of pollen, including the geographical location of the plant, weather and soil conditions, the year of the harvest as well as genetic varieties of plant species.2 In another study it was shown that the Amb a 1 content of short ragweed (Ambrosia artemisiifolia) pollen deviates up to 10-fold when comparing pollen harvested at the same location but in different years.3 Consequently, the extent of batch-to-batch variability allowed in allergen products is significantly greater than, e.g. for chemically derived medicinal products. Furthermore, quality control is complicated for many AIT products marketed in the EU because they are not native extracts but chemically or physically modified. In these cases, many analytical methods cannot be performed at the level of the final product, but are limited to intermediates. Therefore, special regulatory requirements for such products were implemented in recent years. Last but not least, the vast number of allergenic materials had to be taken into account when defining the regulatory framework for allergen product quality control. This includes some special regulations for rare allergens as well as for allergens from closely related species. This review article summarizes the current quality requirements arising from European legislation or European regulatory documents and how the indicated characteristics of AIT products have been accounted for.
Regulatory backgroundAllergen products have been subjected to European pharmaceutical legislation in 1989, when Directive 89/342/EEC extended the Directives 65/55/EEC and 75/319/EEC, including additional provisions for immunological medicinal products consisting of vaccines, toxins or serums and allergens. The directive defined that an “allergen product shall mean any medicinal product which is intended to identify or induce a specific acquired alteration in the immunological response to an allergizing agent”. This definition remained unchanged in the subsequent Directive 2001/83/EC. Importantly, Directive 2001/83/EC states that all medicinal products, either prepared industrially or manufactured by a method involving an industrial process, are required to receive marketing authorization before distribution in a Member State. Despite this requirement, there exist many national exceptions, leading to a highly heterogeneous regulatory situation for allergen products in the EU.4,5 Especially the distribution of so-called “named patient products” (NPP; medicinal products manufactured for an individual patient) according to an exemption as stated in the Directive 2001/83/EC is still very common in many member states, although this exemption from marketing authorization was originally included in the Directive to enable the treatment of rare allergies using customized products. To counteract the distribution of AIT products containing highly prevalent allergens under the guise of NPPs, the Therapy Allergy Ordinance (TAV) was implemented in Germany in 2008.1,6–8 The transition phase is still ongoing. In parallel, various other Member States have established different national strategies to facilitate market access of allergen products.4 In order to counteract this regulatory heterogeneity, a draft reflection paper on harmonization of regulatory procedures for allergen products in the EU was presented to the Coordination Group for Mutual Recognition and Decentralized Procedures – Human (CMDh) in November 2015.9 Subsequently, a drafting group was established with the objective to propose harmonized regulatory approaches for allergen products in the EU. This process is ongoing and no results of the work of this drafting group have been published so far.
The central requirements for quality control of allergen products are obligatorily laid down in the European Pharmacopoeia (Ph. Eur.) in the Monograph on Allergen Products.10 After some extensive revisions in the past, the most recent changes to the monograph account for the establishment of two recombinant major allergen reference standards11,12 as well as the publication of several separate source material monographs like the Monograph on Pollens for Allergen Products.13 The requirements of the Ph. Eur. are mirrored in the Guideline on Allergen Products: Production and Quality Issues (EMEA/CHMP/BWP/304831/2007), complemented with more detailed information in accordance with the current state of knowledge.14 For example, this Guideline introduced the concept of homologous groups to allergen product regulation.15 This concept created a new basis for data extrapolation between allergen groups justified by sound scientific criteria. Meeting the needs of a field with such a wide variety of active substances, data on safety and efficacy can be extrapolated to a certain extent within a homologous group if products fulfil specific criteria.16 However, extrapolation of quality data is limited to stability data as well as process validation.14
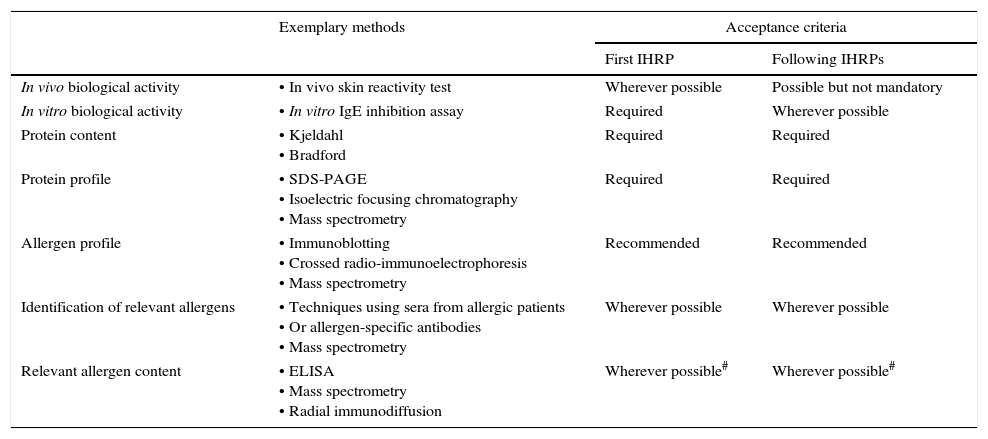
Quality requirements for allergen extractsThe in-house reference preparationThe centre of Ph. Eur. conform allergen extract quality control is the so-called in-house reference preparation (IHRP). For each allergen product, a representative preparation is selected and used to control batch-to-batch consistency at the level of active substance as well as intermediates, and if possible at finished product level. An IHRP should be produced using the same manufacturing process as the corresponding product, with the exception that IHRPs are to be formulated and stored to ensure their stability, which is commonly achieved by lyophilization. Before reaching the end of the shelf-life, a new IHRP has to be implemented relative to the previous IHRP. The characterization steps required by the Ph. Eur. for IHRPs are summarized in Table 1. It should be noted though that the requirements may vary depending on the respective allergenic source, the knowledge on its allergenic components and the prevalence of the allergy. For example, the Monograph requests that the first IHRP is standardized in vivo if a sufficient number of patients sensitized to a given allergen is available. In the EU this standardization procedure is usually performed according to the so called Nordic Guideline, established by the Nordic Council of Medicines,17 but the alternative method described by Turkeltaub is also accepted.18 20 patients sensitized to the allergen in question undergo skin prick testing to enable the comparison of several dilutions of the IHRP with a histamine control. This way, the biological potency of the IHRP can be expressed in biological units.19 By comparing each new IHRP with the previous IHRP in several qualitative and quantitative assays, including an in vitro biological activity assay, all reference preparations and therefore all released batches of an allergen product can be linked back to the first IHRP standardized in vivo. In order to avoid shifting of quality parameters over time, trending analysis has to be performed and a correlation factor has to be established if results of the new IHRP deviate from the previous preparation.
Quality requirements for in-house reference preparations for AIT products.
| Exemplary methods | Acceptance criteria | ||
|---|---|---|---|
| First IHRP | Following IHRPs | ||
| In vivo biological activity | • In vivo skin reactivity test | Wherever possible | Possible but not mandatory |
| In vitro biological activity | • In vitro IgE inhibition assay | Required | Wherever possible |
| Protein content | • Kjeldahl • Bradford | Required | Required |
| Protein profile | • SDS-PAGE • Isoelectric focusing chromatography • Mass spectrometry | Required | Required |
| Allergen profile | • Immunoblotting • Crossed radio-immunoelectrophoresis • Mass spectrometry | Recommended | Recommended |
| Identification of relevant allergens | • Techniques using sera from allergic patients • Or allergen-specific antibodies • Mass spectrometry | Wherever possible | Wherever possible |
| Relevant allergen content | • ELISA • Mass spectrometry • Radial immunodiffusion | Wherever possible# | Wherever possible# |
In accordance with the Monograph on Allergen Products, the IHRP is used as comparator in several quality control tests in order to ensure batch-to-batch consistency of allergen extracts.
Tests to be performed for quality control of allergen extract intended for AIT are listed in Table 2.
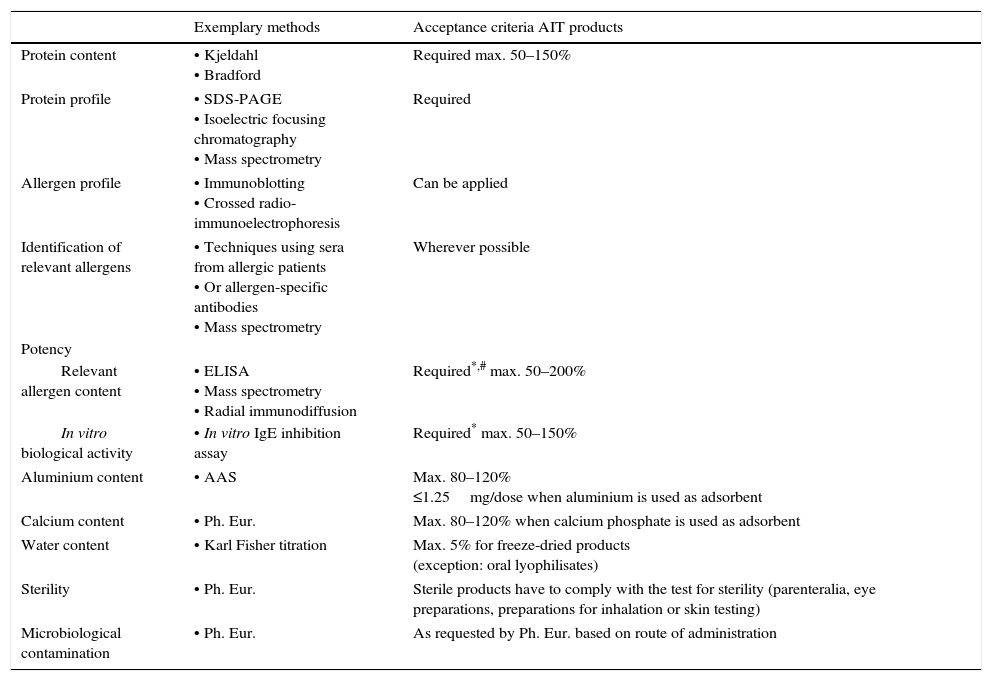
Quality requirements for allergen extracts intended for AIT.
| Exemplary methods | Acceptance criteria AIT products | |
|---|---|---|
| Protein content | • Kjeldahl • Bradford | Required max. 50–150% |
| Protein profile | • SDS-PAGE • Isoelectric focusing chromatography • Mass spectrometry | Required |
| Allergen profile | • Immunoblotting • Crossed radio-immunoelectrophoresis | Can be applied |
| Identification of relevant allergens | • Techniques using sera from allergic patients • Or allergen-specific antibodies • Mass spectrometry | Wherever possible |
| Potency | ||
| Relevant allergen content | • ELISA • Mass spectrometry • Radial immunodiffusion | Required*,# max. 50–200% |
| In vitro biological activity | • In vitro IgE inhibition assay | Required* max. 50–150% |
| Aluminium content | • AAS | Max. 80–120% ≤1.25mg/dose when aluminium is used as adsorbent |
| Calcium content | • Ph. Eur. | Max. 80–120% when calcium phosphate is used as adsorbent |
| Water content | • Karl Fisher titration | Max. 5% for freeze-dried products (exception: oral lyophilisates) |
| Sterility | • Ph. Eur. | Sterile products have to comply with the test for sterility (parenteralia, eye preparations, preparations for inhalation or skin testing) |
| Microbiological contamination | • Ph. Eur. | As requested by Ph. Eur. based on route of administration |
The requirement of a mandatory determination of potency in products intended for AIT can be fulfilled either by determination of total allergenic activity or control of the content of individual relevant allergen molecules. The choice of method is of special importance because the majority of AIT products in the EU is standardized to potency, thus defining the labelled strength on the basis of arbitrary biological units and not on mass units of individual allergen molecules.4 One has to keep in mind though that the degree of correlation between total allergenic activity and therapeutic efficacy still remains unknown for AIT, which is not surprising given that also the mode of action is still not fully understood. In addition, the inherent differences in dosing between AIT with sublingual products and subcutaneous products have to be considered. While the arbitrary units used for expression of total allergenic activity do not allow general statements on optimal dosage, it has been postulated for many years that a specific amount of a major allergen (5–20μg) are the optimal dose to reach a clinically relevant effect without causing unacceptable side effects.20 However, clinical studies using recombinant allergens have shown that the effective dose for purified allergens is potentially higher and greatly product-dependent. For example, studies with a subcutaneously applied hypoallergenic variant of the major birch allergen Bet v 1 identified 80μg as optimum dose for AIT.21 Another phase II study reporting significant effects used 15μg of either recombinant wild type Bet v 1 or natural purified Bet v 1 in monthly injections.22 Moreover, rBet v 1 in sublingual tablets in a dose range of 12.5–50μg was reported to induce a reduction in symptom score compared to placebo irrespective of the applied dose.23 In contrast, a development programme on a cocktail of five recombinant allergens from Phleum pratense with a dose of up to 120μg is reported not to be pursued because promising results from early clinical trials could not be confirmed in a Phase III field trial.24 These examples clearly show that each AIT product, irrespective of its route of application as well as its standardization to total allergenic activity or individual allergen molecules, has to be assessed individually for its safety and efficacy.
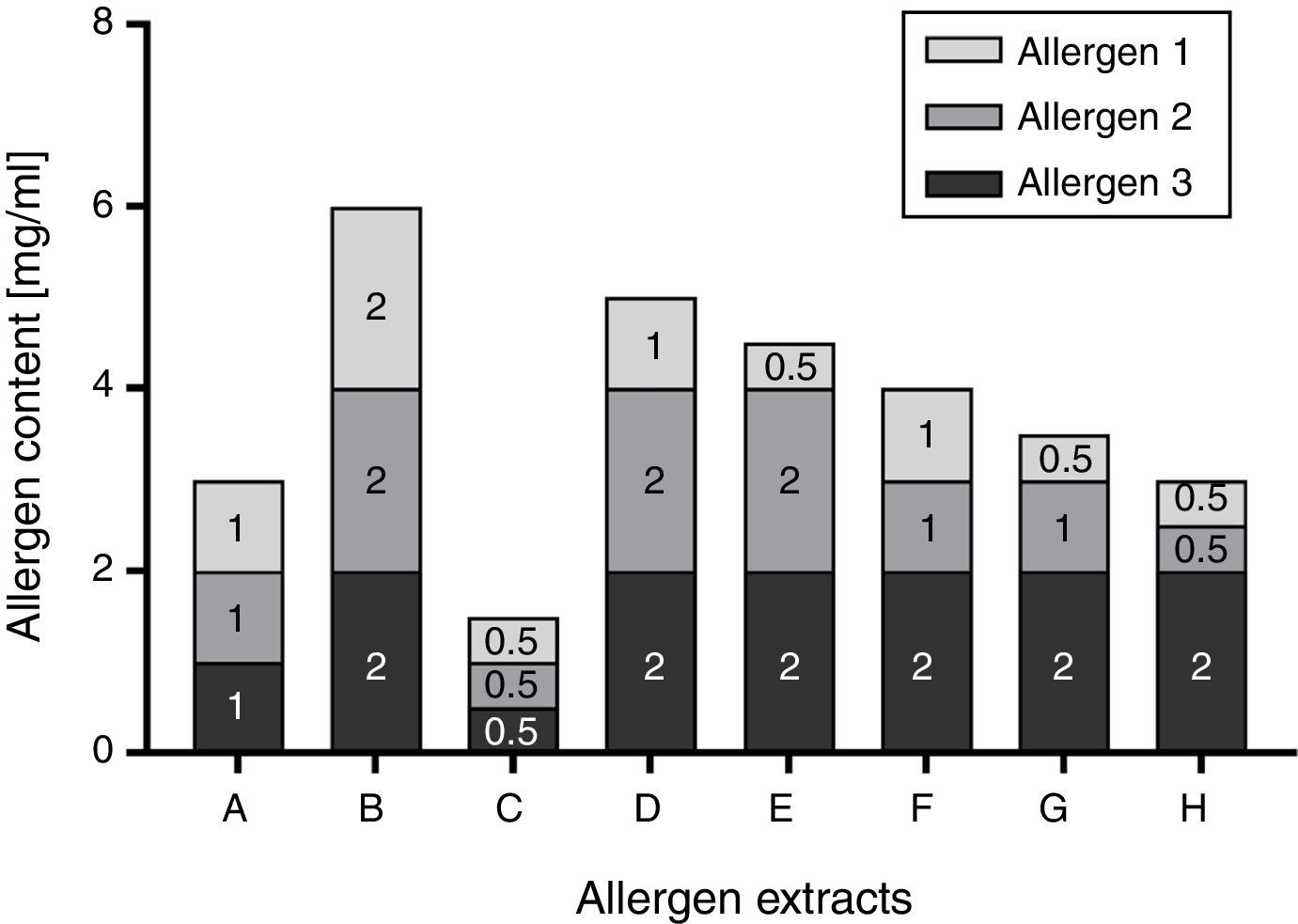
Total allergenic activity is determined based on the IgE-binding capacity of an allergen extract, relative to an IHRP. The main reagent is a pool of sera collected from patients allergic to the allergenic source material in question. Although the preparation of such a pool is regulated by the Guideline on Allergen Products: Production and Quality Issues,14 results of the assay are highly dependent on the selected patient collective, determining the specificities and amount of allergen-specific IgEs in the pool. In addition, the patient sera pool represents a limited resource which needs to be replaced in certain intervals. This limitation could be circumvented by the determination of individual allergen molecules. Allergen molecules are up to now usually quantified in extracts by immunoassays such as allergen-specific enzyme-linked immunosorbent assays (ELISAs). Especially when using monoclonal antibodies, availability of the critical assay components is theoretically unlimited once established. However, the challenge of individual allergen quantification lies in the number of relevant allergens that need to be monitored. Especially for allergen sources with a high number of major allergens, it is certainly ambitious to establish one immunoassay per allergen. In addition, the Guideline on Allergen Products: Production and Quality Issues requests that a correlation between total allergenic activity and the content of relevant allergens has to be demonstrated during development14 in case that the allergen product is defined on the basis of relevant allergens. Consequently, a manufacturer would have to establish an assay for total allergenic activity at some point, even if it is ultimately not used in batch release. While e.g. correlation between total allergenic activity and Bet v 1 content is a well-established fact for birch pollen extracts,25–27 the demonstration of correlation becomes more complex when several major allergens have to be considered. In contrast to total allergenic activity though, potency determination based on major allergen content provides detailed information on the relevant allergen molecules and the ratios between these. As for each relevant allergen specific acceptance criteria need to be established, this may make it more difficult to fulfil all of these and may therefore potentially lead to an increase in the number of batches that have to be rejected at release, even though the approach permits more room for fluctuations because each individual allergen molecule can vary from 50 to 200% of a stated amount (Fig. 2).
 is displayed. In this theoretical example, the stated amount of each allergen was selected to be 1mg/ml. Consequently, the content of each allergen could vary between 0.5 and 2mg/ml, resulting in a potential 4-fold difference in allergen content between allergen extract B and C.")
Schematic illustration of Ph. Eur. conform allergen extract composition based on three allergens. The possible composition of exemplary allergen extracts in accordance with the requirement of the Monograph on Allergen Products (50–200% of the stated amount) is displayed. In this theoretical example, the stated amount of each allergen was selected to be 1mg/ml. Consequently, the content of each allergen could vary between 0.5 and 2mg/ml, resulting in a potential 4-fold difference in allergen content between allergen extract B and C.
An advantage of standardization based on individual allergen molecules is the potential to label allergen extracts in comparable mass units like μg allergen/dose. This would enable inter-product comparison and represent a clear improvement compared to the currently used arbitrary, manufacturer-specific units. However, it has been shown that comparability of results is only possible if the combination of the same reference standard and the same immunoassay is used for analysis.28 So far, two recombinant allergen reference standards have been established,11,12 but the corresponding immunoassays are not yet available as officially approved standard methods.26,29,30 All in all, despite the various assets and drawbacks of each method, the majority of AIT products currently marketed in the EU is standardized based on total allergenic activity. It should be noted though that the content of individual relevant allergen molecules is commonly determined in parallel in many products as part of batch release. However, the information which allergen molecules are controlled by the manufacturers to ensure batch-to-batch consistency of a product is usually not available to physicians or patients.
Quality requirements for chemically modified/adsorbed allergen productsThe requirements of the Monograph on Allergen Products are applicable to native as well as modified allergen extracts. Yet, several tests listed in Table 2 cannot be performed after modification of an allergen extract. Generally, two types of modification are distinguished: Physical and chemical modification. The most common physical modification is the adsorption onto carriers like aluminium hydroxide, calcium phosphate or tyrosine, aiming at modulation of immunological characteristics of an allergen extract. Chemical modification of allergen extracts usually includes conjugation and/or crosslinking with e.g. glutaraldehyde, or carbamylation with cyanates. These modifications aim at destruction of B cell epitopes to reduce IgE-reactivity while retaining immunogenicity due to intact T cell epitopes. The resulting preparations are commonly referred to as allergoids and have been developed to reduce side effects in AIT.31,32
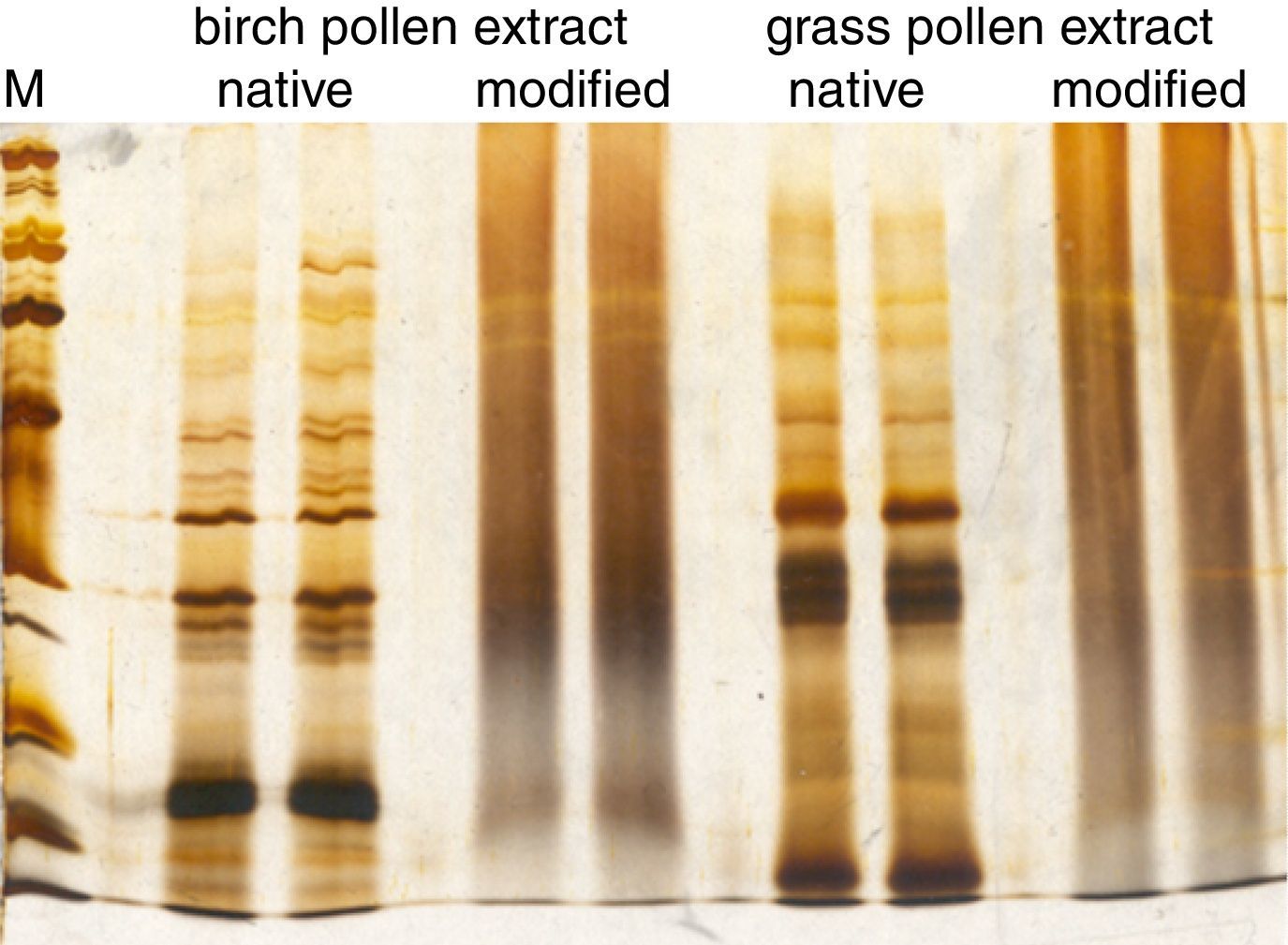
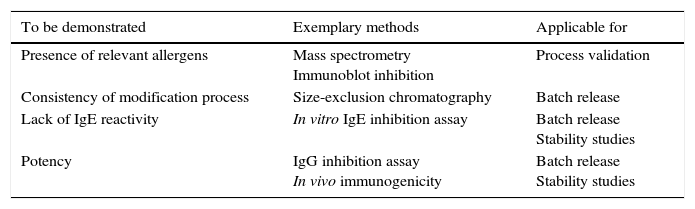
When controlling the quality of allergoids after their chemical modification, several tests laid down in the Monograph on Allergen Products may no longer be performed or have limited significance. For example, the protein profile analyzed by SDS-PAGE after conjugation with glutaraldehyde is reduced to a smear and distinct bands are no longer visible (Fig. 3). Consequently, in cross-linked preparations, the protein profile of a grass pollen allergoid cannot be distinguished from a birch pollen allergoid. Hence, the protein profile by SDS-PAGE is neither suitable to control identity of the product nor the presence of relevant allergen bands. For the same reasons, determination of allergen profiles via Western Blot analysis is usually not possible after chemical modification. Similarly, the determination of total allergenic activity in allergoids is limited to the control of residual potency because by definition and as confirmed by respective testing IgE-reactivity is significantly reduced after chemical modification. Also measuring individual allergen content is not possible in allergoids by way of immunoassays. As a consequence, special requirements for quality control of modified allergen products have been implemented in the regulatory framework. Firstly, it is defined both by the Monograph on Allergen Products as well as by the Guideline on Allergen Products: Production and Quality Issues that all tests for allergen product quality control have to be performed as late as possible in the manufacturing process. Secondly, the amount of total soluble protein and/or the presence of IgE-binding components in the supernatant have to be determined in adsorbed products in batch release as well as in stability studies in order to confirm efficacy and stability of the adsorption. Thirdly, the Guideline requests several additional tests to ensure the consistent quality of modified allergen extracts (Table 3).
 ranges from 6.5kDa to 200kDa.")
Comparison of allergen extract protein profiles before and after chemical modification. SDS-PAGE was performed under reducing conditions with a birch pollen extract and a grass pollen extract before and after chemical modification with glutaraldehyde. The molecular weight marker (M) ranges from 6.5kDa to 200kDa.
Additional quality requirements for allergoids.
| To be demonstrated | Exemplary methods | Applicable for |
|---|---|---|
| Presence of relevant allergens | Mass spectrometry Immunoblot inhibition | Process validation |
| Consistency of modification process | Size-exclusion chromatography | Batch release |
| Lack of IgE reactivity | In vitro IgE inhibition assay | Batch release Stability studies |
| Potency | IgG inhibition assay In vivo immunogenicity | Batch release Stability studies |
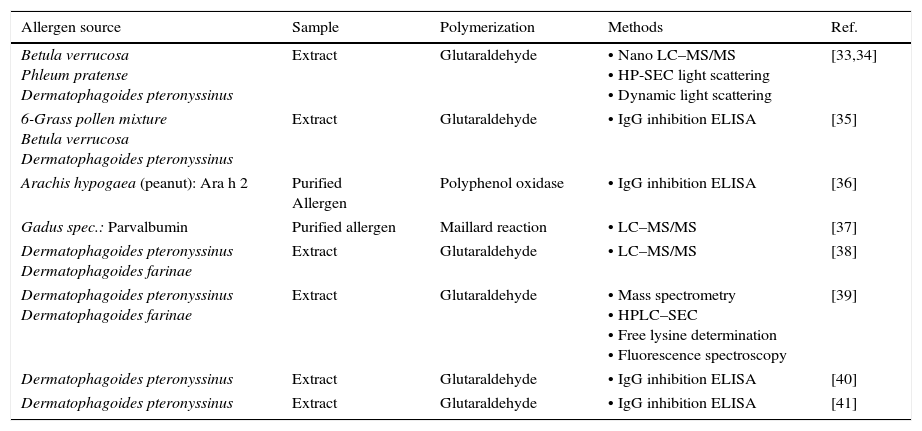
Despite the widespread use of allergoids in the EU, the various products available and the obligation to follow the requirements of the Guideline on Allergen Products: Production and Quality Issues, the number of publications on the topic of allergoid characterization is comparatively low (Table 4). Notably, more than half of the listed publications have been published in “Arbeiten aus dem Paul-Ehrlich-Institut”.
Methods for characterization of chemically modified allergens and extracts published until 2016 (reprint from [4]).
| Allergen source | Sample | Polymerization | Methods | Ref. |
|---|---|---|---|---|
| Betula verrucosa Phleum pratense Dermatophagoides pteronyssinus | Extract | Glutaraldehyde | • Nano LC–MS/MS • HP-SEC light scattering • Dynamic light scattering | [33,34] |
| 6-Grass pollen mixture Betula verrucosa Dermatophagoides pteronyssinus | Extract | Glutaraldehyde | • IgG inhibition ELISA | [35] |
| Arachis hypogaea (peanut): Ara h 2 | Purified Allergen | Polyphenol oxidase | • IgG inhibition ELISA | [36] |
| Gadus spec.: Parvalbumin | Purified allergen | Maillard reaction | • LC–MS/MS | [37] |
| Dermatophagoides pteronyssinus Dermatophagoides farinae | Extract | Glutaraldehyde | • LC–MS/MS | [38] |
| Dermatophagoides pteronyssinus Dermatophagoides farinae | Extract | Glutaraldehyde | • Mass spectrometry • HPLC–SEC • Free lysine determination • Fluorescence spectroscopy | [39] |
| Dermatophagoides pteronyssinus | Extract | Glutaraldehyde | • IgG inhibition ELISA | [40] |
| Dermatophagoides pteronyssinus | Extract | Glutaraldehyde | • IgG inhibition ELISA | [41] |
Especially the development of IgG inhibition ELISAs for allergoid potency determination has to be regarded as a significant improvement compared to immunogenicity testing of allergoids in vivo. The monitoring of allergen specific IgG production in animals is generally conflicting with the 3R principles,42 especially because allergoid potency has to be controlled in each batch produced. In addition, it has been shown that results of in vivo immunogenicity tests with allergoids are commonly highly variable43 and therefore often not sufficiently indicative of allergoid stability. This gap in allergoid quality control is narrowed by in vitro IgG inhibition ELISAs. Typically, such assays are requested to be able to detect a 50% loss of potency in allergoids. However, it needs to be mentioned that such IgG assays may not be suitable for stability testing of adsorbed finished products in all cases.
ConclusionQuality control in agreement with the current regulatory requirements is of major significance to ensure safety and efficacy of AIT products. It is important to note that the use of biological source materials of complex composition in production of AIT products has shaped these quality requirements, representing a balance between the desired level of product-specific standardization on the one hand and the necessary flexibility for certain test parameters on the other hand. In addition, special regulatory requirements have been established, e.g. for allergoids, meeting the need of valid quality control even after modification of an allergen extract. Despite the uniform legal basis provided by the Directive 2001/83/EC, the regulatory status of AIT products remains heterogeneous in the EU member states. All the same, the increasing regulatory requirements provide the necessary basis to ensure AIT product quality. Furthermore, joint activities in the EU drive the establishment of allergen reference standards and corresponding standard methods for individual allergen quantification, because the currently used arbitrary potency units for AIT products do not allow cross-product comparability. In summary, the continuous development of regulatory requirements for allergen extracts has initiated remarkable advancements over the last years and will be further optimized to ensure quality, safety and efficacy of AIT products throughout the EU.
Conflict of interestThe Doctors Zimmer and Bonertz declare that they have no competing interests in relation to this publication and neither financial nor personal relationships with the pharmaceutical industry or other parties that could inappropriately influence their work.
The Dr. Vieths reports personal fees from Ärzteverband Deutscher Allergologen, personal fees from Swiss Society for Allergy and Immunology, personal fees from Schattauer Allergologie Handbuch, personal fees from Elsevier Nahrungsmittelallergien und Intoleranzen, personal fees from Karger Food Allergy: Molecular Basis and Clinical Practice, non-financial support from German Research Foundation, non-financial support from European Directorate for the Quality of Medicines and Health Care, non-financial support from European Academy of Allergy and Clinical Immunology, non-financial support from German Chemical Society (GDCh), non-financial support from AKM Allergiekongress, non-financial support from International Union of Immunological Societies, personal fees from University Hospital Gießen/Marburg, personal fees from University of Bonn, personal fees from Pharmacon, personal fees from Medical University of Vienna, Austria, personal fees from Gesellschaft für pädiatrische Allergologie und Umweltmedizin, non-financial support from World Allergy Organization, non-financial support from Technical University of Munich, non-financial support from Austrian Society for Dermatology and Venerology, outside the submitted work.
ContributionsSV, AB and JZ have designed the structure, drafted and critically revised as well as approved the final version of this review article.
The authors would like to thank Dr Frank Führer, Dr Detlef Bartel, Dr Uwe Müller and Dr Sybille May (Paul-Ehrlich-Institut, Langen, Germany) for provision of SDS-PAGE results.
