








Puntos clave

Los errores innatos del metabolismo (EIM) son enfermedades debidas a un defecto genético, del que resulta una ausencia/anormalidad de una enzima o su cofactor que conduce al déficit o acumulación de uno o varios metabolitos. Fisiopatológicamente se han distinguido 3 tipos:
Tipo intoxicación: la enzima deficiente origina una disminución de la metabolización de un sustrato que, por sí mismo o debido a que se metaboliza por otras vías, causa síntomas de intoxicación aguda o crónica. Suele haber situaciones desencadenantes como estrés físico, infecciones intercurrentes, ayuno o ingesta excesiva de nutrientes que no pueden ser metabolizados.
Tipo déficit energético: la enzima deficiente está implicada en la producción de energía, con el consiguiente déficit funcional.
Acumulación de sustancias complejas que no pueden ser metabolizadas: en este grupo, los síntomas son progresivos y no se relacionan con desencadenantes como las comidas o el ayuno.
Es fundamental para el diagnóstico y el tratamiento precoz un alto grado de sospecha clínica.
EtiologíaLos EIM son producidos por una mutación genética que origina cambios estructurales o funcionales en proteínas codificadas por el gen implicado. Dicha mutación puede causar un déficit enzimático que conlleve acumulación del sustrato, producción de metabolitos anormales y reducción del producto. Otras veces, la proteína implicada se encarga del transporte transmembrana y se da un déficit de la sustancia que debería ser transportada. Por último, el cambio estructural de la proteína puede conllevar un aumento de la actividad enzimática y un exceso de producto1,2.
Las mutaciones pueden surgir de novo o ser heredadas de los padres, en cuyo caso la herencia puede ser de cualquier tipo: autosómica dominante, autosómica recesiva, ligada al sexo y mitocondrial3.
ClínicaLa presentación clínica de los EIM puede afectar a todos los órganos y sistemas. Las manifestaciones clínicas más frecuentes son las neurológicas y las digestivas4 (tabla 1).
Síntomas clínicos más frecuentes que pueden presentar los errores innatos del metabolismo según el órgano afectado
| Neurológicos Coma Apnea/hiperventilación Síntomas psiquiátricos Convulsiones Retraso mental Macrocefalia/microcefalia Hipotonía Miopatías Leucodistrofias Polineuropatías Síndrome cerebeloso Sordera neurosensorial Regresión neurológica Enfermedades hereditarias degenerativas Autoagresividad Afección extrapiramidal |
| Hematológicos Citopenias Hemorragias Trombosis |
| Respiratorios Neumopatía Estridor |
| Digestivos Dolor abdominal Vómitos Esteatohepatitis Problemas de alimentación Diarrea Síndrome de Reye Hepatomegalia Insuficiencia hepática Insuficiencia pancreática exocrina Colestasis Cirrosis |
| Oculares Retinitis pigmentaria Atrofia óptica Mancha rojo cereza Estrabismo Ptosis Ectopia lentis Cataratas |
| Cardíacos Arritmias Defectos de conducción Insuficiencia cardíaca Cardiomiopatías |
| Óseos Alteraciones de columna Hiperlaxitud Fracturas Crisis óseas Artritis Signos de depósito Epífisis punteadas Retracciones articulares Osteoporosis |
| Cutáneos Hiperqueratosis Ictiosis Fotosensibilidad Hipertricosis Alopecia Anomalías del cabello Angioqueratosis Nódulos subcutáneos Lipomas |
| Endocrinos Diabetes Hipotiroidismo Hipogonadismo |
| Renales Tubulopatía Litiasis Poliquistosis Síndrome nefrótico Síndrome hemolítico urémico |
| Otros Muerte súbita, episodios aparentemente letales Deshidratación Abortos Infecciones de repetición Inmunodeficiencias Desnutrición Fallo del crecimiento Dismorfias malformaciones Esplenomegalia |
El ayuno, el estrés, la fiebre, las infecciones intercurrentes, las cirugías, el ejercicio físico y ciertos alimentos o medicamentos pueden desencadenar el inicio de los síntomas o empeorar el cuadro clínico de un EIM5. Por ejemplo, la presentación típica de los trastornos del ciclo de la urea o las acidemias orgánicas es neonatal, aguda y grave, caracterizada por letargia, anorexia, vómitos y shock. Sin embargo, formas más leves pueden presentarse en niños mayores o adultos con episodios de vómitos y alteración del nivel de conciencia, fallo de medro, intolerancia a las proteínas, convulsiones o alteraciones psicomotrices. Algunas enfermedades están relacionadas con la alimentación como la galactosemia (galactosa, lactosa) o la intolerancia hereditaria a la fructosa (fructosa, sacarosa y sorbitol). Arbitrariamente, vamos a dividirlos según la edad de presentación en 3 grupos.
Período prenatal y neonatalInicio prenatal- —
Aborto
- —
Hidrops fetalis: en enfermedades lisosomales (gangliosidosis GM1, galactosialidosis, mucolipidosis II, Gaucher, Pompe, Niemann Pick, mucopolisacaridosis I, VII y IV y otras), anemias y miocardiopatías de origen metabólico.
- —
Defectos del crecimiento y/o morfogénesis: pueden presentar un cuadro sindrómico (Smith-Lemli-Opitz o Zellweger); una macrocefalia puede ser secundaria a un EIM (aciduria glutárica tipo I); madres con hiperfenilalaninemia no diagnosticada pueden tener hijos con una embriopatía3.
- —
Convulsiones intrauterinas.
- —
Enfermedades maternas, como el síndrome HELLP (hemólisis, aumento de enzimas hepáticas y trombocitopenia), pueden deberse a EIM del metabolismo del feto (déficit de 3OH-acil-CoA deshidrogenasa de cadena larga, déficit de CPT I en el feto) y defectos de la cadena respiratoria mitocondrial3.
- —
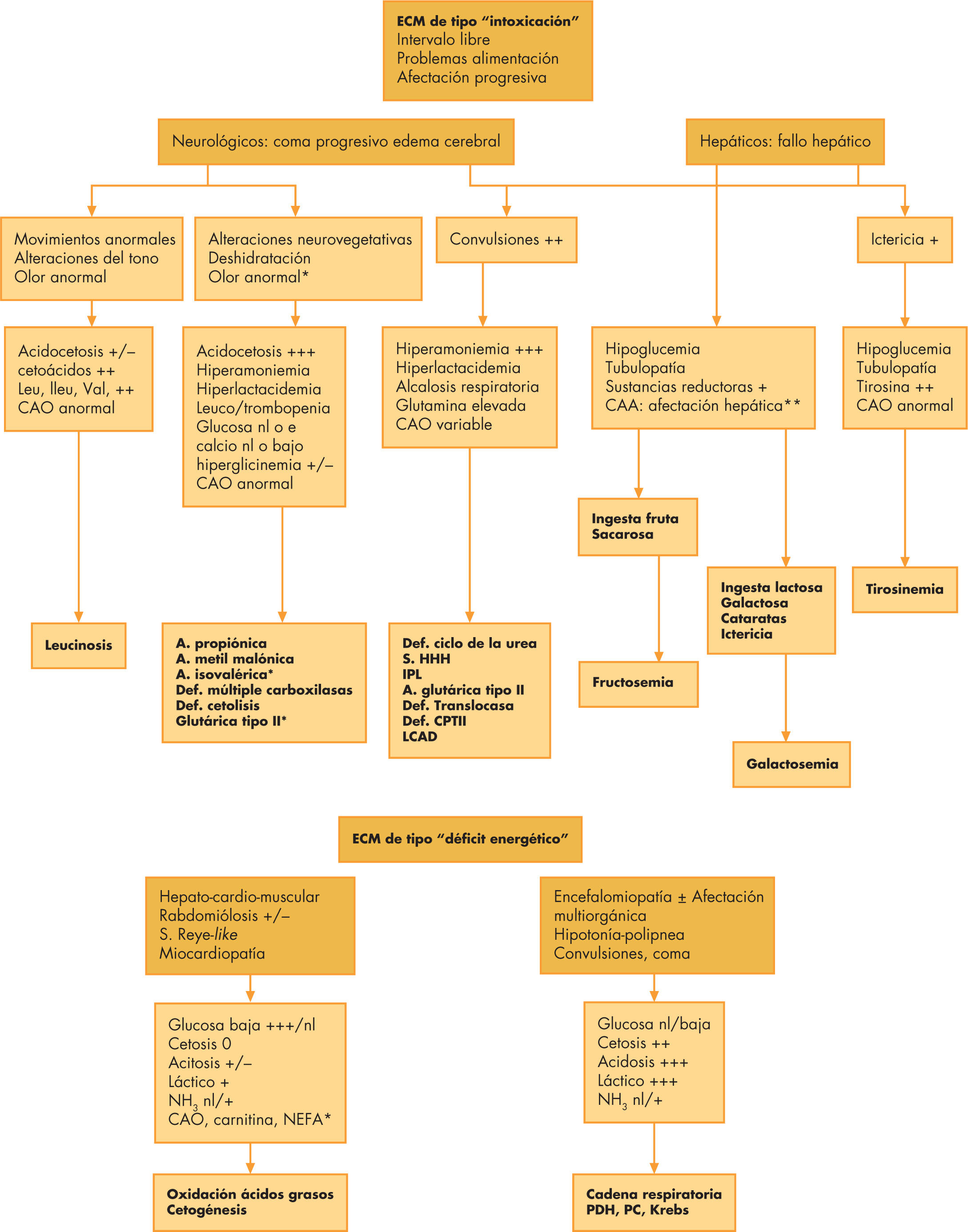
Síntomas encefalopáticos. Según Saudubray et al, hay 2 tipos de cuadros encefalopáticos: “tipo intoxicación” y “tipo déficit energético” (fig. 1). En los EIM del “tipo intoxicación”, por defectos en el catabolismo de los aminoácidos y otras acidosis orgánicas, es típico que haya un “intervalo libre de enfermedad” que oscila entre 1 y 10 días; inicialmente suelen presentarse síntomas digestivos y/o respiratorios o deshidratación, con letargia que progresa hasta un coma. En el “tipo déficit energético” son frecuentes las crisis convulsivas, encefalopatía e hipotonía. Entre las encefalopatías que cursan con convulsiones, es típica la asociación de hiperglicinemia no cetósica y “brote-supresión” en el electroencefalograma (EEG)6. Siempre se deben tener presentes las causas cuyo tratamiento precoz condiciona la evolución de la enfermedad: las convulsiones sensibles al ácido folínico, las sensibles a piridoxina, el déficit en biotinidasa, defectos en la síntesis de serina, defectos del trasporte de glucosa al sistema nervioso central (transportador GLUT1) y déficit de creatina cerebral y todos los ECM que cursen con crisis de hipoglucemia.
 Figura 1.
Figura 1.Diagnóstico diferencial entre los diferentes errores congénitos del metabolismo que cursan con síntomas de tipo intoxicación y de tipo déficit energético. AGII: aciduria glutárica tipo II; CAO: ácidos orgánicos; CPTII: carnitinpalmitoil transferasa; IPL: intolerancia a proteínas con hiperlisinuria; LCAD: déficit de acil-CoA deshidrogenasa; LCHAD: déficit de 3-hidroxiacil-CoA deshidrogenasa; NEFA: ácidos grasos libres no esterificados; PC:piruvato carboxilasa; PDH:piruvato deshidrogenasa; S. HHH: síndrome hiperamoniemia, hiperornitinemia, homocitrulinuria.
(0.55MB). - —
Recién nacido hipotónico: de origen central, neuromuscular (miopatías o polineuropatías) o en cuadros sindrómicos como el síndrome de Prader Willi o cromosomopatías.
- —
Diferentes cuadros en función del órgano afectado. En el período neonatal es frecuente la afección hepática. El fallo multiorgánico es relativamente frecuente en los EIM.
- —
Signos de enfermedad de depósito como anomalías óseas junto con hepatomegalia o esplenomegalia y síntomas hematológicos con anemia o trombocitopenia.
- —
Síntomas transitorios (Niemann Pick tipo C puede cursar con un cuadro colestásico en el período neonatal). Las enfermedades mitocondriales también pueden cursar con síntomas que remitan transitoriamente (convulsiones, pausas de apnea, alteraciones cardíacas).

- —
Alteraciones neurológicas: retraso psicomotor, hipotonía, regresión neurológica, convulsiones, síntomas neurológicos deficitarios transitorios, afección cerebelosa, piramidal o extrapiramidal, leucodistrofias o dismielinizaciones, neuropatías y miopatías.
- —
Síndrome de Reye o similar a éste.
- —
Síndrome de muerte súbita del lactante o episodios con riesgo vital7.
- —
Síntomas digestivos: vómitos, disfunción hepática, fallo de medro, ictericia, etc.8. Algunos síntomas aparecen en relación con el tipo de alimentación, como la intolerancia hereditaria a la fructosa y la galactosemia.
- —
Otros síntomas (tabla 1).
El diagnóstico diferencial entre las diferentes enfermedades se realiza como en el apartado anterior. Los cuadros neurológicos que se instauran en este período suelen ser regresivos.
Exámenes complementarios y diagnósticoLa historia clínica es la herramienta fundamental para el diagnóstico: síntomas y signos de la enfermedad, relación con el ayuno, ingesta de ciertos alimentos o medicamentos, antecedentes familiares (un árbol genealógico familiar detallado debe constar siempre en la historia clínica), etc.
Entre los exámenes complementarios necesarios, están los incluidos en la tabla 24,9–11. Es importante destacar algunos aspectos:
- —
Distinguir el tipo de presentación del EIM orienta el proceso diagnóstico.
- —
Las muestras deben ser recogidas en el momento agudo, ya que así aportan una información más fidedigna. No obstante, no se debe anteponer la recogida de muestras a la estabilización del paciente.
- —
Si no supone riesgo de descompensación, generalmente las pruebas deben realizarse en ayunas. Sin embargo, para los EIM tipo déficit energético es recomendable el estudio del estado de oxidación-reducción en ayunas y posprandial.
- —
Si no van a ser enviadas a un laboratorio especializado inmediatamente, se debe guardar en congelador las muestras de suero, plasma, orina u otros fluidos. Las muestras de tejidos deben ser congeladas a temperaturas más bajas (–80°C). Si se quiere realizar estudio enzimático en fibroblastos, dicha muestra nunca debe congelarse.
- —
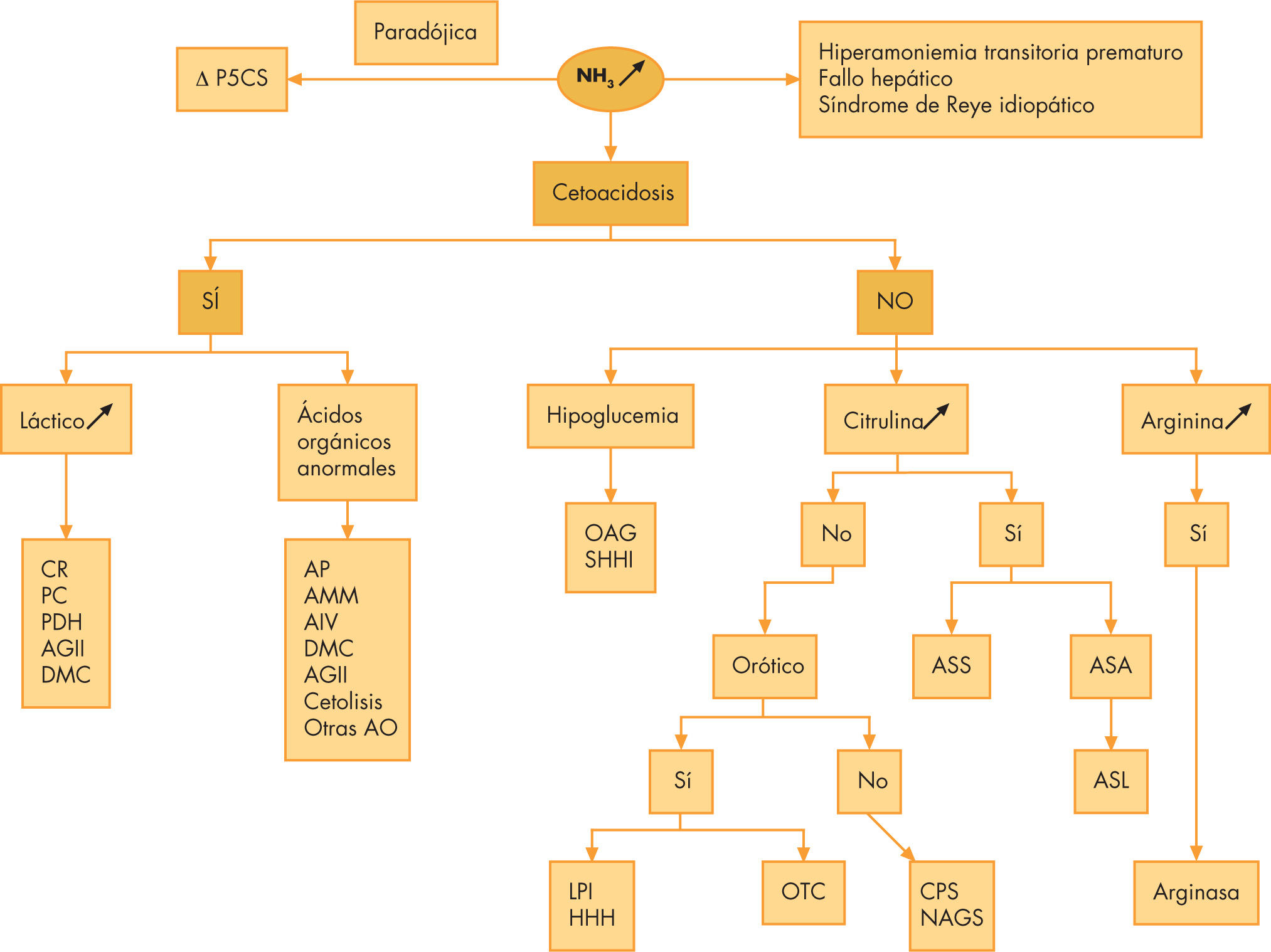
La hiperamoniemia (fig. 2) orienta hacia un EIM en muchas ocasiones y la determinación de amonio debe ser incluida en el proceso diagnóstico de todo neonato grave o con sospecha de sepsis. Sin embargo, también pueden verse cifras elevadas de amonio en la insuficiencia hepática o en enfermedades graves.
 Figura 2.
Figura 2.Diagnóstico diferencial de las hiperamoniemias. AGII: aciduria glutárica tipo II; AS: argininosuccinato sintetasa; ASA: ácido argininosuccinico; ASL: argininosuccinato liasa o argininosuccinasa; ASS: argininosuccinato sintetasa; CPS: carbamilfosfato sintetasa; CR: cadena respiratoria mitocondrial; DMC: déficit múltiple de carboxilasas; ∆ P5CS: delta pirrolina 5 carboxilato sintetasa; HHH: hiperamoniemia, hiperornitinemia, homocitrulinuria; LPI: intolerancia a las proteínas con hiperlisinuria; NAGS: N-acetilglutamato sintetasa; OAG: oxidación de los ácidos grasos mitocondrial; OTC: ornitintranscarbamilasa; PC:piruvato carboxilasa; PDH piruvato deshidrogenasa; SHHI: hiperinsulinismo, hipoglucemia, hiperamoniemia de la infancia.
(0.21MB). - —
La hipoglucemia, por sus graves consecuencias neurológicas, debe ser estudiada en todos los pacientes salvo en los casos de hipoglucemia precoz y transitoria en el período neonatal.
- —
En caso de dismorfias, se deben valorar estudios de mucopolisacáridos y oligosacáridos en orina, esteroles, transferrina deficiente en hidratos de carbono y/o ácidos grasos de cadena muy larga.
- —
En el diagnóstico diferencial de hepatopatías, es conveniente incluir el estudio de ácidos biliares, además de los estudios basales. Ante la sospecha de galactosemia, se debe retirar la galactosa, y ante la sospecha de fructosemia, se debe evitar la ingestión de fructosa, sacarosa y sorbitol. Debe valorarse la posibilidad de enfermedad de Niemann-Pick tipo C.
- —
Si el paciente tiene convulsiones, es importante recoger una muestra de líquido cefalorraquídeo para estudio de neurotransmisores y aminoácidos, así como practicar una determinación de sulfitos en orina. Se debe intentar un ensayo terapéutico con piridoxina, ácido folínico y piridoxal-fosfato.
- —
Estudios post mórtem: son fundamentales en los casos de muerte súbita o cuando la rapidez en la evolución de la enfermedad no ha permitido descubrir su etiología. Para ello se requiere protocolizar la recolección, el procesamiento y almacenaje de muestras post mórtem. Luego se dirigirán los estudios dependiendo del cuadro clínico y los datos necrópsicos4,12,13. Las muestras que deben recogerse son: suero, plasma, líquido cefalorraquídeo y orina, que deben congelarse de inmediato; muestras de sangre total con heparina o EDTA (que hay que congelar) y sangre en papel de Guthrie; muestras de tejidos: músculo e hígado, que se congelan de inmediato en nitrógeno líquido para estudios enzimáticos; biopsia de piel, que se debe realizar en condiciones de esterilidad: el fragmento de 3-4mm de diámetro se pone en un frasco con suero fisiológico a temperatura ambiente para realizar cultivo de fibroblastos; y necropsia completa.

Exámenes de laboratorio en pacientes con sospecha de error congénito del metabolismo
| Estudios basales |
| Análisis de sangre Hemograma, coagulación, ionograma, calcio, gasometría (brecha aniónica), glucosa, perfil hepático, ácido úrico, colesterol Amonio, ácidos láctico y pirúvico, cuerpos cetónicos, ácidos grasos no esterificados (NEFA) Otros (según clínica y sospecha diagnóstica): enzimas musculares, etc. |
| Análisis de orina Orina elemental: glucosa, pH, cuerpos cetónicos Sustancias reductoras Test de sulfitos (Sulfitest) Electrolitos, ácido úrico, creatinina |
| Estudios metabólicos |
| Análisis de sangre: se efectúan en plasma, suero, sangre en papel de Guthrie, sangre con ácido etilendiaminotetracético (EDTA) (según enfermedad). Los más utilizados son los siguientes: Estudio del estado de oxidación-reducción célular*: lactato/piruvato, 3OH-butirato/acetoacetato, NEFA/cuerpos cetónicos*, glucosa por cuerpos cetónicos* Aminoácidos* Carnitina y acilcarnitinas* Transferrina deficiente en hidratos de carbono Otros: homocisteína, ácidos grasos de cadena muy larga (VLCFA) y esteroles |
| Análisis de orina Aminoácidos* Ácidos orgánicos* Ácido orótico Otros: creatina y guanidinoacético, purinas y pirimidinas, mucopolisacáridos, oligosacáridos, sales biliares, polioles, sulfátidos etc. Otros estudios dependiendo de los síntomas clínicos: líquido cefalorraquídeo para bioquímica* y estudios metabólicos específicos* |
| Estudio de extensión de la enfermedad |
| Exámenes de apoyo Exámenes radiológicos: radiografía de tórax y esqueleto; estudios neurorradialógicos: resonancia magnética con espectroscopia, ecografías Estudio cardiológico Estudio oftalmológico Estudios histoquímicos en tejidos: biopsias de piel, hepática, muscular, etc. |
| Estudios enzimáticos y genéticos Estudios enzimáticos: se utilizará el tejido más accesible que exprese la enzima Estudios genéticos: se precisan muestras que contengan ADN, como sangre en papel de Guthrie, sangre con heparina o EDTA, muestras de tejidos que se congelan |
Individualmente, los EIM son enfermedades infrecuentes, pero en conjunto se estima que 1/500 recién nacidos vivos presenta un EIM.
Los síntomas pueden ser muy variados, dado el gran número de defectos enzimáticos posibles. Pueden afectar a prácticamente todos los órganos y sistemas. Las manifestaciones neurológicas y gastrointestinales son las más frecuentes. Las técnicas de cribado neonatal son muy limitadas y permiten realizar diagnósticos precoces de sólo unas pocas enfermedades, por lo que en los demás casos el diagnóstico se basa en la sospecha clínica y la adecuada orientación de los exámenes complementarios.
El pronóstico depende del diagnóstico, la eficacia del tratamiento existente, la instauración precoz del tratamiento, el estado clínico del paciente cuando se inicie el tratamiento (si hay o no lesiones irreversibles) y el control metabólico en el curso de la enfermedad, entre otros.


