




Puntos clave
- •
El tratamiento actual de las cardiopatías congénitas en la infancia permite que la mayoría de los niños lleguen a la edad adulta.
- •
Las cardiopatías congénitas son muy diversas y tienen un manejo médico, intervencionista y quirúrgico específico e individualizado, que requiere de un equipo especializado y multidisciplinar.
- •
El pediatra que lleva a niños con cardiopatías congénitas debe conocer la entidad, el tratamiento, la situación del niño y sus secuelas, así como las posibilidades futuras de actuaciones terapéuticas para poder anticiparse y detectar los problemas intercurrentes.
- •
La transición del adolescente a edad adulta debe hacerse de una manera progresiva y continua, iniciándose a partir 12 años, para conseguir una implicación progresiva del adolescente en la evolución de su cardiopatía.
- •
Los estudios, la orientación laboral, la actividad física y el deporte, la contracepción y los riesgos de la gestación son temas fundamentales en la transición a la edad adulta.
El tratamiento de las cardiopatías congénitas ha evolucionado en los últimos años de manera espectacular, y en la actualidad el 85% de los niños que nacen con cardiopatías, hace unos años incompatibles con la vida, llegan a la edad adulta1,2. Esto ha supuesto la creación de las Unidades de Cardiopatías congénitas del adulto y la aparición de nuevos retos en el manejo global del paciente con cardiopatía congénita.
Lectura rápida
El tratamiento de las cardiopatías congénitas ha evolucionado en los últimos años de manera espectacular y en la actualidad el 85% de los niños que nacen con cardiopatías, hace unos años incompatibles con la vida, llegan a la edad adulta.
El tipo de cardiopatías congénitas es muy diverso y su tratamiento actualmente se centra en manejo médico, cateterismo intervencionista y quirúrgico.
En la actualidad, la mayoría de las correcciones quirúrgicas se realizan en periodo neonatal. Aunque pueden quedar lesiones residuales o secuelas que requieren cirugías posteriores.
El cateterismo intervencionista hoy en día está solucionando problemas de cardiopatías simples y aisladas y resolviendo las complicaciones que van apareciendo en el seguimiento, condicionando una disminución del número de intervenciones quirúrgicas.
Las cirugías cardiacas en cardiopatías congénitas se dividen en cirugías cerradas (sin necesidad de extracorpórea, habitualmente por toracotomía lateral) y abiertas (con circulación extracorpórea).
Existen algunas cardiopatías congénitas que pueden pasar desapercibidas en edad pediátrica.
Algunos niños con cardiopatías congénitas pueden llegar a la adolescencia sin tratamiento previo, con cirugías paliativas o con reparaciones completas fisiológicas o anatómicas que presentan defectos residuales, secuelas o ambos.
El pediatra o médico de cabecera que lleve al paciente debe de conocer la entidad, las secuelas de estos pacientes o defectos residuales, y si puede requerir cirugía futura.
Existe un número, cada vez más pequeño, de pacientes que no pudieron ser intervenidos en edad pediátrica, o bien su clínica pasó inadvertida y desarrollaron en el seguimiento elevación de la presión pulmonar que condicionó el síndrome de Einsenmenger. En situación de Einsenmenger, los pacientes no pueden ser intervenidos de su cardiopatía de base. Solo el trasplante cardiopulmonar o el trasplante pulmonar con corrección de su cardiopatía, si esta es simple, son las opciones de tratamiento. En la actualidad, con los nuevos fármacos que controlan la presión pulmonar, la supervivencia de los pacientes en Einsenmenger es cada vez mayor.
Los adolescentes con cardiopatías congénitas deben seguir una vida saludable, no fumar, restricción de sodio en comidas, evitar la anemia. Es recomendable ejercicio aeróbico de intensidad de acorde con su situación clínica.
La transición consiste en preparar al paciente en afrontar y conseguir la autogestión de su salud. Para la planificación de una buena transición es fundamental un trabajo conjunto entre las Unidades de Cardiología Pediátrica y de cardiopatías Congénitas del adulto. La transición necesariamente ha de seguir contemplándose de manera multidisciplinar y requiere el concurso de diferentes especialidades médicas, similar a lo acontecido en edad pediátrica.
La adolescencia es la fase de transición entre la infancia y la edad adulta y supone una evolución, tanto física como psicológica, siendo difícil establecer la franja de edad exacta donde se produce la transición. Según la Organización Mundial de la Salud (OMS), la adolescencia se sitúa entre los 10–11 años hasta los 19 años, abarcando el proceso de pubertad y la adquisición de autonomía personal con capacidad, del hasta ahora niño, en la autogestión de su vida y, por tanto, de su salud. En aquellos niños que nacieron con anomalías cardiacas congénitas supone un reto, tanto para ellos y sus familias como para el profesional sanitario, que debe saber realizar una transición adecuada y atender las nuevas necesidades que van apareciendo para conseguir adaptar a estos pacientes a una vida plena.
Hay muchos factores genéticos y ambientales cuya interacción puede alterar la formación del corazón durante las primeras fases del desarrollo fetal (las primeras 8 a 9 semanas de embarazo). A veces se conoce la causa de una cardiopatía congénita y son cada vez más las alteraciones genéticas asociadas a las cardiopatías. La exposición a determinados factores ambientales durante el primer trimestre de embarazo puede provocar anomalías estructurales3,4. Las cardiopatías congénitas tienen una incidencia estimada entre el 5–8 por 1.000 nacidos vivos; de ellas, el 40–60% va a requerir de una intervención a lo largo de su vida. En la actualidad, con los avances en el manejo quirúrgico y el cateterismo intervencionista, así como con el avance en las técnicas diagnósticas y el manejo médico en las Unidades de Críticos Pediátricas, estos niños son intervenidos en edades cada vez más precoces; incluso recientemente, se están realizando técnicas intervencionistas durante la gestación. A lo largo de la vida, estos niños precisarán de nuevas intervenciones, y deben de ser conscientes y tener conocimiento de su patología de base para enfrentarse a los sucesivos tratamientos. En algunas cardiopatías, donde solo se pueden realizar actuaciones paliativas (su corazón, a pesar de las intervenciones nunca va a poder funcionar como el corazón normal) se les opera interponiendo prótesis en posición pulmonar, que deben ser intercambiadas conforme el paciente va creciendo; ello supone irremediablemente sucesivas cirugías con riesgo de fracaso de estas y requerir de un trasplante cardiaco como opción final.
Clasificación de las cardiopatías congénitas y tratamientoLas cardiopatías congénitas se pueden clasificar según una clasificación fisiopatológica o funcional (según la alteración clínica que producen: cianosis, insuficiencia cardiaca o ambas) o una clasificación anatómica. Dentro de esta última, la clasificación más empleada es la del análisis segmentario de Robert Anderson5, donde se analizan las relaciones anatómicas de las venas, con las cavidades cardiacas y los vasos. Existen múltiples y variadas anomalías cardiacas, con posibilidad de combinación de unas con otras, lo que hace que su número se incremente considerablemente. No hay 2 cardiopatías idénticas, pero sí modelos similares. Pueden tratarse de ausencia de un ventrículo, de una válvula aurículo-ventricular, de un vaso, anomalías del drenaje de venas pulmonares o sistémicas, etc., siendo desde anomalías complejas (incluyen varias anomalías anatómicas, derivadas de una misma alteración embriológica) o anomalías simples, como defectos septales o estenosis valvulares aisladas.
El tipo de cardiopatías congénitas es muy diverso y su tratamiento actualmente se centra en el manejo médico, cateterismo intervencionista y quirúrgico. En la actualidad, la mayoría de las correcciones quirúrgicas se realizan en el periodo neonatal. Aunque pueden quedar lesiones residuales o secuelas que requieren cirugías posteriores. El cateterismo intervencionista hoy en día está solucionando problemas de cardiopatías simples y aisladas, y resolviendo las complicaciones que van apareciendo en el seguimiento, condicionando una disminución del número de intervenciones quirúrgicas. Existen dispositivos específicos para cierre de defectos septales, como defectos interauriculares y algunos interventriculares, persistencia de conducto arterioso o fístulas, y se pueden realizar aperturas de vasos estrechos, como en la coartación de aorta o valvuloplastias para las estenosis de válvulas (incluso intervencionismo fetal) y, más recientemente, interposición de stent para apertura de vasos y de válvulas en posición pulmonar y aórtica por cateterismo6.
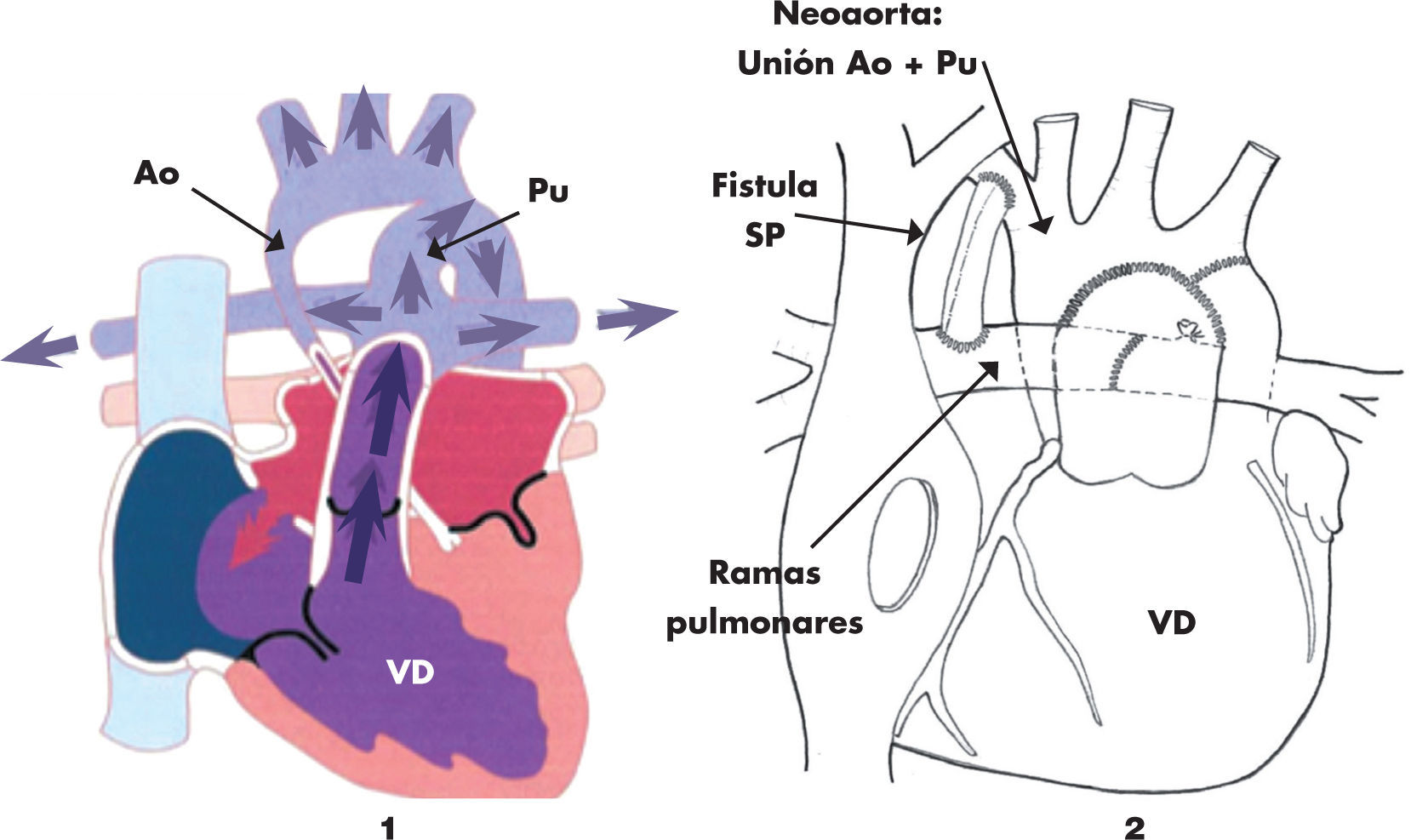
La primera intervención quirúrgica en una cardiopatía congénita se realizó en 1938 en Boston y consistió en el cierre de un conducto arterioso por toracotomía lateral. La aparición, en 1958, del oxigenador de membrana supuso un cambio radical en la cirugía, condicionó el poder «parar» el corazón y mantener al cuerpo oxigenado con la circulación extracorpórea (CEC) mientras se realizaba la intervención. El progresivo desarrollo tecnológico permitió la aparición de la cirugía cardiaca neonatal en los años 80. En 1975, el Dr. Jatene intervino con éxito la corrección anatómica de un niño de un año con una transposición de grandes arterias (inversión en la posición de los vasos, de tal manera que la aorta está conectada con el ventrículo derecho y la pulmonar con el izquierdo), técnica que se empezó a utilizar en neonatos por el Dr. Castañeda a partir de los años 80. De entre todas las técnicas, la de más riesgo y la última en desarrollarse ha sido la cirugía que se realiza en los niños afectados de síndrome de hipoplasia de cavidades izquierdas (SHCI), llamada cirugía Norwood (fig. 1); son recién nacidos que nacen sin desarrollo del ventrículo izquierdo, ni de la aorta; la cirugía consiste en unir la miniaorta y la arteria pulmonar y formar un único vaso, desconectando las ramas pulmonares y manteniendo el flujo a través de un tubo entre ramas pulmonares y ventrículo derecho (o con la aorta dependiendo de la técnica). El desarrollo del diagnóstico prenatal ha disminuido la incidencia del SHCI y los casos que se operan tienen un resultado de supervivencia que varía según el centro entre el 60 y el 80%. En España, el desarrollo en el manejo de las cardiopatías congénitas se debe gracias al Dr. Manuel Quero, del Hospital Ramón y Cajal, pionero durante los años 80. A partir de los años 90, hay un desarrollo de la cirugía cardiaca pediátrica a nivel nacional y todas las técnicas se realizan en España en la actualidad, en un total de alrededor de 20 centros. En el centro del autor, como ejemplo, la cirugía de Jatene se realiza desde 1991, con un número total cercano a los 300 casos intervenidos y una supervivencia actual entre el 92 y el 98% (según la anatomía).
 y la cirugía de Norwood clásico (2). Ao: aorta; Pu: pulmonar; SP: sistémico-pulmonar (de aorta a ramas pulmonares).")
Las cirugías cardiacas en cardiopatías congénitas se dividen en cirugías cerradas (sin necesidad de extracorpórea, habitualmente por toracotomía lateral) y abiertas (con CEC). Habitualmente, se denominan en función del cirujano que realizó la técnica por primera vez. Si el paciente tiene 2 ventrículos de tamaño similar y con buena función, y un vaso de tamaño adecuado, se realiza una cirugía correctora o biventricular (se intentará que el corazón sea lo más parecido al corazón normal), el vaso quedará como aorta y se colocará una prótesis (tubo) en posición pulmonar. Los tubos empleados pueden ser homoinjertos (aorta o pulmonares de cadáveres procesados en un banco de tejidos y desnaturalizados y criopreservados), biológicos (xenoinjerto, de origen animal, o de ingeniería genética) o de material sintético. Todos estos tubos tienen la desventaja de una durabilidad corta y que en el niño pequeño tienen que ser de pequeño tamaño (adecuados a su peso) y deben ser cambiados cuando el niño crece. Lo que es imprescindible para el éxito de la cirugía en general es que las ramas pulmonares tengan un desarrollo adecuado, si existe hipoplasia severa de las ramas pulmonares, no se podrá ofrecer una corrección quirúrgica completa. Las cirugías correctoras biventriculares más frecuentes han sido realizadas en pacientes con tetralogía de Fallot y en los defectos aurículo-ventriculares (típicamente asociado al síndrome de Down) desde los años 70 y 80. Cuando solo existe un ventrículo funcional (bien porque exista una atresia de la válvula aurículo-ventricular, o una ausencia de un ventrículo, o es imposible conectar el ventrículo sistémico con la aorta), a estos pacientes se les realiza una cirugía univentricular (llamada cirugía de Fontan), donde las venas cavas se conectan mediante un tubo (habitualmente de un material protésico de PTLE) con las arterias pulmonares, dejando solo un ventrículo único como corazón.
Otra manera de clasificar a las cirugías es en anatómicas (corrección del defecto según su problema anatómico) o fisiológicas (correcciones quirúrgicas que hacen que el corazón funcione correctamente, aunque no hayamos corregido su defecto anatómico). El ejemplo lo tenemos en la transposición de grandes arterias. Esta cardiopatía condiciona una cianosis intensa (la sangre oxigenada no circula a la aorta). La primera técnica quirúrgica que apareció en los años 70–80, llamada Mustard o Senning, consistía en colocar un parche intraauricular y desviar la sangre que llega de los pulmones y reconducirla al ventrículo derecho; es el llamado switch auricular y consiste en una corrección fisiológica, con esta técnica el paciente deja de estar cianótico pero el ventrículo derecho queda conectado con la aorta, sigue teniendo una cardiopatía, esta vez paliada. En la actualidad esta cirugía ha sido sustituida por la nombrada anteriormente, el switch arterial o técnica de Jatene; se trata de una corrección anatómica: se cambian los vasos de posición (se realiza transferencia de la aorta con sus coronarias) dejando el corazón anatómicamente normal7.
Como vemos, un número cada vez mayor de niños intervenidos con cardiopatías congénitas están en edad adolescente o entrando en edad adulta; se trata de cirugías muy recientes, con supervivencias que han mejorado en los últimos años.
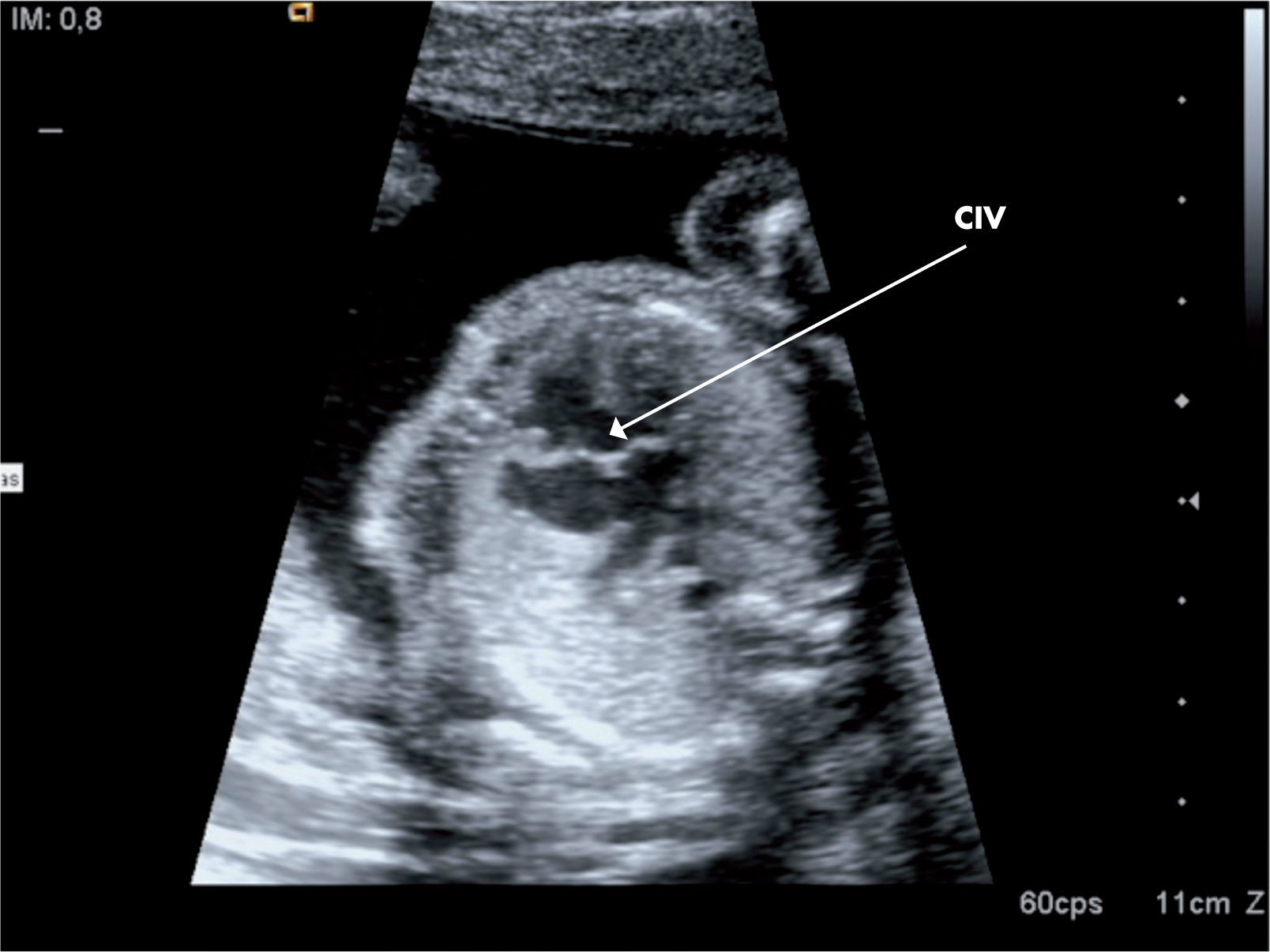
Cardiopatías congénitas no diagnosticadas en edad pediátricaAunque en la actualidad el diagnóstico de cardiopatías congénitas ya se está realizando incluso en periodo fetal (fig. 2), existen algunas cardiopatías congénitas que pueden pasar inadvertidas en edad pediátrica. La sospecha clínica viene dada por la auscultación de un soplo cardiaco de características orgánicas. Entre ellas, las más frecuentes son: la válvula aórtica bicúspide, la estenosis valvular pulmonar, la comunicación interauricular (CIA) tipo ostium secundum, la estenosis subaórtica, conductos arterioso persistente y la coartación aórtica. La válvula aórtica bicúspide puede ser normofuncionante por mucho tiempo. La estenosis pulmonar valvular se suele solucionar con una valvuloplastia pulmonar (mediante cateterismo intervencionista). La CIA genera un cortocircuito izquierda derecha, con sobrecarga de volumen en el ventrículo derecho; el soplo viene determinado por el hiperaflujo a nivel pulmonar. Este defecto puede causar hipertensión pulmonar en la edad adulta si no se diagnostica y se cierra a tiempo. Actualmente, la técnica de elección de cierre es por cateterismo intervencionista, salvo en los defectos muy amplios, donde se procederá a cierre quirúrgico con parche. La estenosis subaórtica, generalmente por un resto muscular llamado rodete, va generando una obstrucción subaórtica progresiva y requiere la extirpación quirúrgica de ese músculo, evitando además la lesión de la válvula aórtica. Pueden pasar inadvertidos conductos arteriosos persistentes, algunos sin repercusión hemodinámica; son los llamados conductos silentes, donde es controvertido su cierre. Si el conducto es sintomático (soplo continuo, dilatación cavidades izquierdas en la ecocardiografía, etc.), debe ser cerrado, la gran mayoría por cateterismo intervencionista; y por último, otra cardiopatía que se puede diagnosticar en el niño mayor, adolescente o adulto es la coartación aórtica; suele diagnosticarse dentro del estudio de una hipertensión arterial sistémica.
.")
Una cardiopatía compleja que puede no dar sintomatología y suele ser de diagnóstico casual en la edad adulta es la transposición corregida de grandes arterias8. Se trata de una inversión ventricular, generada durante el desarrollo embriológico; el «asa ventricular» realiza una rotación hacia la izquierda (asa-L) en vez de hacerlo hacia la derecha (asa-D), como es lo habitual; de modo que el ventrículo morfológicamente derecho se ubica a la izquierda y el izquierdo a la derecha. Como consecuencia, la aorta emerge del ventrículo morfológicamente derecho ubicado a la izquierda y la arteria pulmonar lo hace del ventrículo morfológicamente izquierdo ubicado espacialmente a la derecha. Como está compensada, el paciente puede estar asintomático, pero se asocia característicamente a alteraciones de la conducción aurículo-ventricular y puede presentar un bloqueo aurículo-ventricular en su evolución, así como fallo del ventrículo derecho situado a nivel sistémico. Si se asocia a otras anomalías cardiacas, puede presentar síntomas precoces. En caso de fallo del ventrículo derecho en su evolución a largo plazo, el trasplante cardiaco es una opción óptima cuando falla el tratamiento médico conservador.
Aspectos relevantes en el seguimiento de las cardiopatías congénitas del adolescenteAlgunos niños con cardiopatías congénitas pueden llegar a la adolescencia sin tratamiento previo, con cirugías paliativas o con reparaciones completas fisiológicas o anatómicas que presentan defectos residuales, secuelas o ambos. El pediatra o médico de cabecera que lleve el paciente debe de conocer la entidad, las secuelas de estos pacientes o defectos residuales, y si puede requerir cirugía futura. Es importante estar en contacto con los especialistas que tratan al paciente.
En el seguimiento de los pacientes con cardiopatías congénitas lo más importante que se debe controlar es la aparición de arritmias, derivadas de vías anómalas asociadas a cardiopatías y trastornos de conducción, pero en muchas ocasiones por las propias cicatrices quirúrgicas. En caso de presentar palpitaciones, se deberá realizar un electrocardiograma (ECG) urgente para ver si hay cambios con su ECG basal y solicitar Holter si la sintomatología es frecuente y de larga duración. Un apartado importante es si el paciente presenta un síncope o dolor precordial; determinadas cardiopatías pueden provocar arritmias ventriculares; son síntomas graves que requieren ser revisados urgentemente por los especialistas. En ocasiones, existe un fallo del músculo cardiaco, bien por condicionantes anatómicos (ventrículo derecho en posición anatómica sistémica o ventrículos únicos de morfología derecha) o por fallos tras varias intervenciones, o sobrecargas de volumen o presión (lesiones residuales como regurgitaciones de las válvulas aurículo-ventriculares, insuficiencia aórtica, etc.); en estos casos, cuando falla el manejo médico conservador, se recurre al trasplante cardiaco como opción, aunque las intervenciones previas dificultan la intervención y el resultado a largo plazo.
La endocarditis es otra complicación que se debe tener en cuenta en el seguimiento, aunque con las medidas actuales higiénicas el riesgo es muy bajo. Recientemente, se han cambiado las medidas profilácticas de endocarditis y se es menos tendente a administrar profilaxis9,10.
Existen un número, cada vez más pequeño, de pacientes que no pudieron ser intervenidos en edad pediátrica, o bien su clínica pasó inadvertida y desarrollaron en el seguimiento elevación de la presión pulmonar que condicionó el síndrome de Einsenmenger11. Este consiste en una hiperplasia del músculo del vaso pulmonar, generando elevación fija de las resistencias pulmonares. Si la cardiopatía subyacente es un defecto septal, el cortocircuito izquierda-derecha se invierte, pasando a ser de derecha-izquierda, con lo que el paciente empieza a estar cianótico (coloración azulada de la piel por aumento de la cantidad de hemoglobina reducida en sangre) e incluso puede llegar a estar hipóxico (falta de oxígeno en los tejidos). La hipoxia condiciona un aumento de la producción de eritrocitos a nivel medular, consecuentemente aparece poliglobulia. Según los niveles de hemoglobina, cuando esta es elevada, aparece un aumento de la viscosidad de la sangre que puede condicionar aparición de complicaciones como fenómenos embólicos y abscesos. En situación de Einsenmenger, los pacientes no pueden ser intervenidos de su cardiopatía de base. Es muy importante evitar hipotensión, anemia y otros factores ambientales que pueden desencadenar crisis de hipertensión pulmonar y muerte. Solo el trasplante cardiopulmonar, o el trasplante pulmonar con corrección de su cardiopatía, si esta es simple, son las opciones de tratamiento. En la actualidad, con los nuevos fármacos que controlan la presión pulmonar, la supervivencia de los pacientes con síndrome de Einsenmenger es cada vez mayor. La tasa de sobrevida es del 77% a los 15 años y del 42% a los 25, y las causas de muerte más comunes son muerte súbita (30%), insuficiencia cardiaca (25%) y hemoptisis (15%).
En general, en los adolescentes con cardiopatías congénitas, es importante que lleven una vida saludable, no fumar, restricción de sodio en comidas, evitar la anemia (debe darse hierro si baja la hemoglobina o el volumen corpuscular medio). Es recomendable el ejercicio aeróbico de intensidad de acorde con su situación clínica. Hay que tener en cuenta que una vida sedentaria se ha relacionado con mayor mortalidad que los pacientes con actividad física moderada. Para cardiopatías más complejas, en general, se recomienda ejercicio moderado de tipo aeróbico para todo tipo de niño con cardiopatía, intervenida o no. En este tipo de ejercicio, aunque incremente la carga de volumen, no incrementa la carga de presión. Los ejercicios estáticos o isométricos con resistencia alta y los que provocan aumento de frecuencia cardiaca a niveles máximos (ejercicios de competición) deben de evitarse en determinadas cardiopatías por el riesgo de progresión de su enfermedad (niños con síndrome de Marfan con dilatación de raíz aórtica); por riesgo de arritmias ventriculares; por riesgo de obstrucción al flujo sanguíneo (estenosis aórtica severa, coartación o recoartación aórtica con gradiente diferencial superior a 25 mmHg); por riesgo de crisis de hipertensión pulmonar (síndrome de Einsenmenger) o por muerte súbita (tetralogía de Fallot o estenosis aórtica severa). Para ver su estado físico y su capacidad funcional, se puede realizar prueba de esfuerzos con gases12; las pruebas de esfuerzos estándar (sin gases) se suelen solicitar en el seguimiento para descartar alteraciones hemodinámicas con cambios en la presión arterial o los fenómenos isquémicos.
Transición del adolescente a Unidades de Cardiopatías Congénitas del AdultoMuchos de los adolescentes desconocen el nombre de su cardiopatía, las intervenciones que se les ha realizado, su predisposición a presentar endocarditis bacteriana, el efecto del tabaco y el alcohol en su cardiopatía, cómo detectar síntomas de alarma o de descompensación y la posibilidad de trasmisión de la cardiopatía a sus descendientes. Hasta ahora, la cardiología de las cardiopatías congénitas había sido controlada por los pediatras especializados, pero recientemente, derivado a que un número mayor de niños llegan a edad adulta, es cuando se ha despertado el interés entre los cardiólogos13–16.
La transición consiste en preparar al paciente en afrontar y conseguir la autogestión de su salud. Para la planificación de una buena transición, es fundamental un trabajo conjunto entre las Unidades de Cardiología Pediátrica y de Cardiopatías Congénitas del Adulto. La transición necesariamente ha de seguir contemplándose de manera multidisciplinar y requiere el concurso de diferentes especialidades médicas, similar a lo acontecido en la edad pediátrica.
Muchos de estos niños pueden haber presentado secuelas neurológicas y se ha observado una elevada incidencia de niños con síndrome de hiperactividad17 relacionados, entre otros factores, con las cirugías en periodo neonatal y/o alteraciones del flujo cerebral de la propia cardiopatía. Esto hace que tengan dificultad en concentrarse en los estudios. A su vez, el autoproteccionismo de las familias por su enfermedad hace que puedan ser muy dependientes de sus padres y cuesta que se hagan responsables de su propia enfermedad. Para una transición adecuada, también inciden aspectos laborales y de ocio. Por ello, se debe contemplar la orientación y la preparación para la elección de una adecuada vida laboral. Debe hacerse de forma conjunta con los padres o tutores, en el contexto sociocultural del paciente, en relación con su condición mental y física, y de acuerdo con su capacidad funcional.
Es un hecho que durante el embarazo, el parto y posparto se producen cambios hemodinámicos, especialmente sobrecarga de volumen, que en las pacientes con cardiopatías puede derivar en importantes complicaciones, tanto para las madres como para el feto18,19. La información del riesgo del embarazo debe ser individualizada para cada paciente y de acuerdo con su cardiopatía, a fin de que aprendan a autogestionar su vida sexual y reproductiva, evitando los riesgos que pueda suponer embarazos no planificados o incluso no deseados. Las recientes guías europeas de práctica clínica catalogan a las pacientes en la clasificación modificada de la OMS19 dependiendo del riesgo de complicaciones maternas y fetales; se considera contraindicación para gestación las pacientes con hipertensión arterial pulmonar grave de cualquier causa, las pacientes con disfunción ventricular grave, la presencia de miocardiopatía periparto previa, la presencia de estenosis mitral grave, la presencia de estenosis aórtica grave sintomática, la coartación aórtica nativa grave, o la presencia de síndrome de Marfan con aorta superior a 45mm o de válvula bicúspide con aorta superior a 50mm. Además, debe advertirse con anterioridad sobre la contraindicación de algunos fármacos durante la gestación y su presencia en el tratamiento de las pacientes debería incluirse en la evaluación multidisciplinar de planificación del embarazo.
A pesar de los avances técnicos, los pacientes con cardiopatías congénitas hasta este momento tienen una expectativa de vida por debajo de la población general. Algunos adolescentes no son conscientes de su cardiopatía y esta supone enfrentarse a la información sobre su mortalidad, y puede generar trastornos psicológicos, con periodos de ansiedad y/o depresión o cambios en la personalidad. A su vez, debemos recordar que algunos pacientes presentan otras malformaciones, alteraciones genéticas o lesiones de otros órganos que les hacen ser todavía dependientes de tutores o familiares.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.