




Puntos clave
- •
Es necesaria la investigación clínica (IC) en niños, pero realizada adecuadamente y respetando la dignidad y los derechos fundamentales de los participantes en los procesos de investigación.
- •
El consentimiento informado tras un proceso de información adecuada es imprescindible para la participación de niños en IC.
- •
Se debe ir adaptando la información a la evolución intelectual del menor y utilizar las herramientas adecuadas para que el menor la comprenda.
- •
Cualquier proyecto de investigación clínica debe ser evaluado por un CEIC-CEI.
- •
En el caso de investigaciones que supongan ensayos clínicos o procedimientos invasivos en menores de edad, el promotor o el investigador debe informar al ministerio fiscal.
Desde el punto de vista general, la investigación es el conjunto de actividades encaminadas a desarrollar o contribuir a un conocimiento generalizado. En particular, la investigación clínica (IC) es la actividad investigadora que utiliza seres humanos o material biológico o biográfico procedente de los mismos, y que pretende la comprensión de la enfermedad o el desarrollo de tecnología médica útil para el diagnóstico, la prevención o el tratamiento de la misma. En este sentido, la sociedad considera que la IC es fundamental para una correcta asistencia a los ciudadanos.
Sin embargo, hubo una época en que existió un abuso en la utilización de menores en experimentos científicos, seguida de otra en la que se consideró a los niños sujetos a una especial protección, dando lugar a una restricción a la IC en niños1.
En la actualidad, este cambio pendular ha sido detenido: es necesaria la IC, pero realizada adecuadamente y respetando la dignidad y los derechos fundamentales de los participantes en los procesos de investigación, sobre todo en el caso de los niños.
Necesidad de la investigación clínica en niñosEn el caso del uso de medicamentos en niños, hasta bien entrado el siglo XXI, gran parte de los medicamentos utilizados en niños no habían sido probados en ellos, considerándose fuera de indicación (off-label). En adultos, es excepcional esta situación. Según distintos datos, la proporción de fármacos que se utilizan fuera de indicación es alrededor del 90% en Unidades de Cuidados Intensivos Neonatales, el 70% en Intensivos Pediátricos, el 50% en Digestivo, el 47% en Cardiología y el 36% en Pediatría General2. Este hecho ha sido denominado por algunos «orfandad terapéutica».
Además de lo expuesto sobre medicamentos y ensayos clínicos (EC), han surgido otras líneas de IC relacionadas con pruebas y procedimientos diagnósticos y terapéuticos invasivos, utilización de muestras biológicas, estudios genéticos y estudios observacionales, que son fundamentales para la obtención de conocimientos y su traslación a la clínica y, por lo tanto, capaces de modificar la evolución y pronóstico de las enfermedades pediátricas.
Es obvio que la investigación en niños presenta una serie de problemas relacionadas con las características de un organismo en crecimiento y desarrollo, tipo de enfermedades, número de pacientes y aspectos éticos derivados de su vulnerabilidad.
Pero no es menos cierto que no investigar en ellos conduce a una falta de conocimientos para aplicar en el diagnóstico y tratamiento de sus enfermedades, por lo que los convertimos de nuevo en pacientes más vulnerables ante las enfermedades. La especial protección que deben tener los niños no implica la exclusión de los mismos en aquellas investigaciones relacionadas con las enfermedades propias de este periodo de la vida3.
Ante este problema, las autoridades sanitarias de la Unión Europea adoptaron medidas con objeto de estimular los EC con medicamentos en los niños.
El 26 de enero del 2007 entra en vigor el Reglamento n.° 1901/2006 del Parlamento Europeo y del Consejo, de 12 de diciembre del 2006, sobre medicamentos de uso pediátrico. Este documento enmarca un conjunto de acciones encaminadas a promover el uso seguro y eficaz de medicamentos en pediatría. Para obtener estos objetivos, deben realizarse EC en esta etapa de la vida, teniendo en cuenta la complejidad añadida propia de una época en desarrollo, que incluyen momentos evolutivos tan heterogéneos como es la época de recién nacido, la primera infancia o lactancia o la época puberal y de la adolescencia4.
Todo esto ha llevado a la necesidad de aceptar la IC en niños por considerarlos una población específica, con enfermedades propias de la edad teniendo en cuenta los criterios de beneficio, riesgo y el grado de vulnerabilidad.
Principios éticos que se deben tener en cuenta en la investigación clínicaVista la importancia que tiene la IC en niños y el reconocimiento social de la misma, no solo se puede, sino que se debe hacer. Para que no se repitan los desgraciados hechos del pasado con experimentos abusivos y falta de respeto a los derechos de los participantes, la investigación debe cumplir con una serie de principios éticos y legales. Aunque la IC en menores debe respetar los mismos principios éticos y normas legales que rigen, en general, para las investigaciones en adultos, hay que tener en cuenta además la situación de especial vulnerabilidad que se da en esta época de la vida.
Los principios éticos a considerar en cualquier tipo de investigación biomédica o clínica se fundamentan en una serie de códigos éticos que se desarrollaron a partir del juicio de Nuremberg (1945–1946). Surgen tras la divulgación de las condiciones en las que se realizaron investigaciones abusivas para las personas. Estos son el Código de Nuremberg5, de 1947, la Declaración de Helsinki, en 1964, con actualizaciones periódicas hasta Brasil6, en 2013, el Informe Belmont7, de 1979, Convenio de Oviedo8, de 1997, y la Declaración Universal sobre Bioética y Derechos Humanos de la UNESCO9, de 2005.
A partir de estos códigos y declaraciones, se desarrollan los 4 principios actuales de la bioética, que no solo ponen el marco ético para la investigación, sino que también constituyen un instrumento para evaluar decisiones clínicas desde el punto de vista ético.
Aunque no son derechos, normas o mandatos definitivos, como no podía ser de otra forma, las diversas normativas de muchos estados los han ido incorporando a la legislación sobre IC. Son principios que aun cuando no estén exentos de controversias o limitaciones, constituyen un marco ético universal y coherente10.
En la actualidad, hay unanimidad en que cualquier tipo de investigación que se haga en seres humanos, deberá ajustarse a los llamados grandes principios de la Bioética, con las especiales características de la población a la que nos estamos refiriendo. Estos principios son:
- 1.
Principio de no maleficencia: nos obliga a retirar, prevenir o evitar el daño intencionado, físico, psíquico, moral o social. El principio de no maleficencia, que en su inicio estaría incluido dentro del principio de beneficencia, daría relevancia a la obligatoriedad de no producir daño durante la investigación. En concreto, valorando la relación riesgo-beneficio. En este sentido, es habitual catalogar el nivel de riesgo como «riesgo mínimo», «ligero aumento sobre el riesgo mínimo» y «riesgo mayor que el mínimo».
El «riesgo mínimo» es aquel similar al que puede tener un niño en su vida diaria o durante la realización de un examen físico o un test psicológico rutinario.
Así se entiende que la realización de IC que no supongan un riesgo mayor que el mínimo es bien aceptada. Sin embargo, cuando hay un incremento de riesgo sobre el mínimo se tiende a valorar su realización teniendo en cuenta:
- –
La perspectiva de obtener beneficios directos por su participación y que superen a los del riesgo.
- –
La perspectiva de obtener conocimientos generalizados importantes para la población, aunque no haya posibles beneficios y siempre y cuando el riesgo fuera similar al que tendría en una situación médica parecida.
No deberían programarse IC con incremento importante de riesgo y sin perspectivas de beneficio directo.
- –
- 2.
Principio de justicia.: obliga al reparto equitativo de beneficios y cargas. El principio de justicia referido a la distribución equitativa de beneficios y cargas derivados de la investigación entre toda la población. No se puede ni se debe investigar solo en determinados estratos de población o en países débiles, o en determinados grupos sociales con características de vulnerabilidad.
- 3.
Principio de autonomía: obliga a respetar y promover las decisiones de la persona en la gestión de su salud. Este principio se plasma en la práctica mediante la obtención del consentimiento informado. Se debe resaltar que para tener valor, sobre todo moral, ha de basarse en una información veraz, comprensible, adecuada y adaptada a la persona a la cual se informa. Debe evitarse tanto la falta de información como el exceso de ella.
La obtención del consentimiento para participar en una IC es uno de los temas más importantes, no en vano es la expresión explícita de la autonomía de la persona. Debe considerarse como un proceso que se inicia con la información y termina con la aceptación al otorgar el consentimiento para participar en la IC propuesta. No puede haber un acto de consentir libre y autónomo si la información recibida no es adecuada. Por esto mismo, el proceso de informar ha de hacerse con objetividad, claridad, veracidad, y estar seguro que la persona que recibe la información la ha comprendido de tal manera que pueda formarse su juicio y decidir su participación. El investigador adquiere un gran compromiso en este acto y es su responsabilidad hacer que la información no tenga sesgos que inciten a la participación mediante una influencia indebida. Para ello deberá adaptar la información al nivel de comprensión que observe tanto en los responsables del niño como en el niño y ser capaz de responder a sus dudas. Sobre todo, deben dar explicaciones muy claras de los riesgos y los potenciales beneficios.
Es importante tener presente que, aunque los responsables del niño tienen que dar el consentimiento hasta que el paciente tenga 18 años para participar en un EC, la normativa específica que no deberá incluirse a un niño en un EC si este no da su asentimiento. El asentimiento es un acto imperfecto de aceptación realizado por una persona parcialmente incapaz, en este caso por ser menor; se entiende por tal el acuerdo positivo del niño, y tiene gran valor moral. La capacidad del menor de dar su asentimiento depende no solo de la edad, sino de otros factores, como el grado de madurez, la capacidad intelectual y el tipo de enfermedad. Se considera que por debajo de los 3 años de edad es prácticamente imposible obtener el asentimiento.
Dentro de unos parámetros razonables, debe seguirse un criterio favorable a la consideración de que la madurez del menor es suficiente como para que participe en las decisiones que le atañen, siendo informado, opinando o incluso decidiendo.
- 4.
Principio de beneficencia. obliga a hacer el bien para el paciente, teniendo en cuenta lo que él considera como tal por sí mismo. El principio de beneficencia, por el cual se debe conseguir el bienestar de las personas que participan en una investigación y procurar el máximo de beneficios con el mínimo daño como consecuencia de las intervenciones realizadas en el curso de la investigación.
Los Comité Ético de Investigación Clínica-Comité de Ética de la Investigación (CEIC-CEI) son los órganos independientes, de composición multidisciplinar, constituidos por profesionales sanitarios y no sanitarios, encargados de velar por la protección de los derechos, seguridad y bienestar de los sujetos que participen en la investigación biomédica y además dar garantía pública de ello.
Además de los principios señalados, no deberíamos olvidar el principio de precaución. Puede inscribirse en la ética de la responsabilidad, en tanto considera los efectos de las acciones de los hombres de hoy sobre las generaciones futuras. Este principio demanda el ejercicio activo de la duda, la incertidumbre, aquello que se puede temer sin poder ser evaluado.
Aspectos legales y tipos de investigaciónLa Constitución española promueve y garantiza la investigación científica en el ámbito del interés general. Pero también establece unos límites en la protección de la juventud y la infancia que en ningún caso pueden extralimitarse. Aunque ya hemos dicho que la IC en menores debe respetar las mismas normas legales que rigen en general para las investigaciones en adultos, el hecho de ser vulnerables, y más en dependencia de sus distintas etapas de desarrollo, ha hecho que dichas normas tengan distintos mecanismos de protección. Esto se materializa sobre todo en la prohibición de que corran determinados riesgos que puedan perjudicar su desarrollo, así como en la previsión de que otros sustituyan su capacidad de decidir, sin tener en cuenta el beneficio del menor.
Así, para proteger la integridad física, la normativa dice que no es lícito someter a un sujeto a riesgos más allá de los mínimos, si es que no se espera un beneficio directo para la salud. Por lo tanto, la investigación solo se llevará a cabo en enfermedades pediátricas con objeto de mejorar el bienestar de dicha población.
En el aspecto de la capacidad de decidir, el ordenamiento jurídico establece limitaciones referidas a distintos tramos de edad, lo que supone que hasta que no se haya alcanzado una edad determinada, existe un defecto de capacidad que se suple mediante la patria potestad o tutela. Sin embargo, en la actualidad se reconoce la madurez del menor, para que pueda participar en las decisiones que le atañen, debiendo ser informado, opinando o incluso decidiendo11. En otras palabras, el menor debe dar el asentimiento y tener en cuenta su opinión antes de iniciar su participación.
El marco legal referente a la IC en España es amplio. Hay una legislación muy garantista para los sujetos participantes.
Entre las principales normas que tenemos, debemos citar a la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios12, y su desarrollo mediante el RD 223/2004 que regula los EC con medicamentos13. Recoge una especial protección de los sujetos participantes, especialmente cuando se trata de menores e incapacitados. Así señala que se obtendrá el consentimiento informado previo de los padres o del representante legal del menor; el consentimiento deberá reflejar la presunta voluntad del menor y podrá retirarse en cualquier momento sin perjuicio alguno para él. Cuando el menor tenga 12 o más años, deberá prestar además su consentimiento para participar en el ensayo.
Otra norma que afecta a cualquier tipo de investigación es la Ley 15/1999 y su reglamento posterior que protege la recogida y el tratamiento de los datos personales de los sujetos participantes14,15.
Debido al incremento y la complejidad de otro tipo de investigaciones no relacionadas con los EC con medicamentos, se dictó la Ley 14/2007, que regula la investigación biomédica relacionada con la obtención de muestras, su almacenamiento, investigación con células, embriones o células embrionarias, terapia celular, estudios genéticos e investigación de procedimientos invasivos16.
Por último, no se debe olvidar en este grupo de normas con carácter general a la Ley 41/2002, básica, reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica. Podríamos considerarla una ley transversal que influye y afecta a la normativa de cualquier tipo de investigación17.
Además de esta normativa que podríamos llamar general, existen otras de desarrollo más específicos, aplicables a los distintos tipos de investigación.
En la actualidad podríamos diferenciar distintos tipos de investigación a los que se aplicaría una reglamentación transversal o general y otra más específica en dependencia del tipo. Estos serían:
- 1.
EC con medicamentos. EC es toda investigación efectuada en seres humanos para determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de detectar las reacciones adversas, y/o estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia.
Antes de realizar un EC, es necesaria la obtención del dictamen favorable del Comité Ético de Investigación Clínica (CEIC) correspondiente, la conformidad de la dirección de los centros en donde vaya a realizarse y la autorización de la Agencia Española del Medicamento y Productos Sanitarios (AEMPS). En caso de EC multicéntricos hay un dictamen único que lo realiza un CEIC de referencia. Salvo excepciones reguladas por la norma, todo EC requiere la contratación de un seguro de responsabilidad civil. En los ensayos con menores de edad, es obligatorio informar al ministerio fiscal.
- 2.
IC con productos sanitarios. Este tipo de investigación se efectúa sobre cualquier instrumento, dispositivo, programa informático, material u otro artículo, utilizado solo o en combinación, incluidos los programas informáticos destinados por su fabricante a finalidades específicas de diagnóstico y/o terapia y que intervengan en su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos con fines de diagnóstico, prevención, control, tratamiento o alivio así como investigación, sustitución o modificación de la anatomía de un proceso fisiológico.
Al igual que en el caso anterior, para su inicio debe tener el dictamen favorable del CEIC correspondiente. Es necesaria la contratación de un seguro de responsabilidad civil si el producto sanitario no tiene la marca de la Comunidad Europea.
- 3.
IC con implantes de células, tejidos y órganos. Aunque no existen normas específicas respecto a la IC sobre trasplante de órganos, hay que tener en cuenta que este tipo de investigación solo puede realizarse en centros autorizados para estas actividades. Requieren el dictamen del CEIC del centro coordinador del proyecto, la autorización de los responsables de los centros y la autorización de la autoridad competente de la comunidad autónoma correspondiente.
- 4.
Estudios con procedimientos invasivos. Un procedimiento invasivo en investigación de define como toda intervención realizada con fines de investigación que implique un riesgo físico o psíquico para el sujeto afectado.
Para poder iniciar un estudio de este tipo es necesaria la obtención del dictamen favorable del CEIC del centro correspondiente y la autorización por la autoridad competente de la comunidad autónoma. Este procedimiento exige de la contratación de una póliza de seguro. Asimismo también hay que informar de la realización del estudio al ministerio fiscal.
- 5.
Estudios postautorización de tipo observacional. Pueden ser con medicamentos o con productos sanitarios. En ambos casos, la asignación de un paciente a una terapéutica concreta no estará decidida de antemano como en un protocolo de EC, sino que estará determinada por la práctica habitual de la medicina, y la decisión de prescribir un medicamento determinado estará claramente disociada de la decisión de incluir al paciente en el estudio.
Este tipo de estudio requiere el dictamen de un único CEIC acreditado en España.
- 6.
Estudios de investigación con procedimientos no invasivos. Investigación con datos de carácter personal. Este grupo incluye a todos aquellos estudios que no han quedado reflejados en los grupos descritos anteriormente. Es un grupo difícil de definir y no hay una normativa específica que los regule, aunque pueden ser aplicables los principios éticos y normas legales de las disposiciones generales referidas en los anteriores grupos. No obstante, se recomienda que soliciten la evaluación por parte de un CEIC acreditado y se informe de su realización en el centro correspondiente.
- 7.
Estudios en los que se obtengan muestras biológicas. Análisis genéticos. Cada día es más frecuente que en cualquiera de los tipos de investigación citados arriba se extraigan muestras biológicas como parte de los objetivos del estudio. Estas muestras pueden ser utilizadas para los objetivos de la investigación o ser almacenadas en forma de colecciones o biobancos.
Con respecto a la investigación y los análisis genéticos, es importante recordar lo que dice la norma sobre el límite a los mismos: solo podrán hacerse pruebas predictivas de enfermedades genéticas o que permitan identificar al sujeto como portador de un gen responsable de una enfermedad, o detectar una predisposición o una susceptibilidad genética a una enfermedad, con fines médicos o de investigación médica y con un asesoramiento genético, cuando esté indicado, o en el caso del estudio de las diferencias interindividuales en la respuesta a los fármacos y las interacciones genético-ambientales o para el estudio de las bases moleculares de las enfermedades.
Por lo tanto, conviene tener en cuenta la aplicación de la normativa cuando en cualquiera de los tipos de investigación citados se manejen muestras biológicas y datos genéticos.
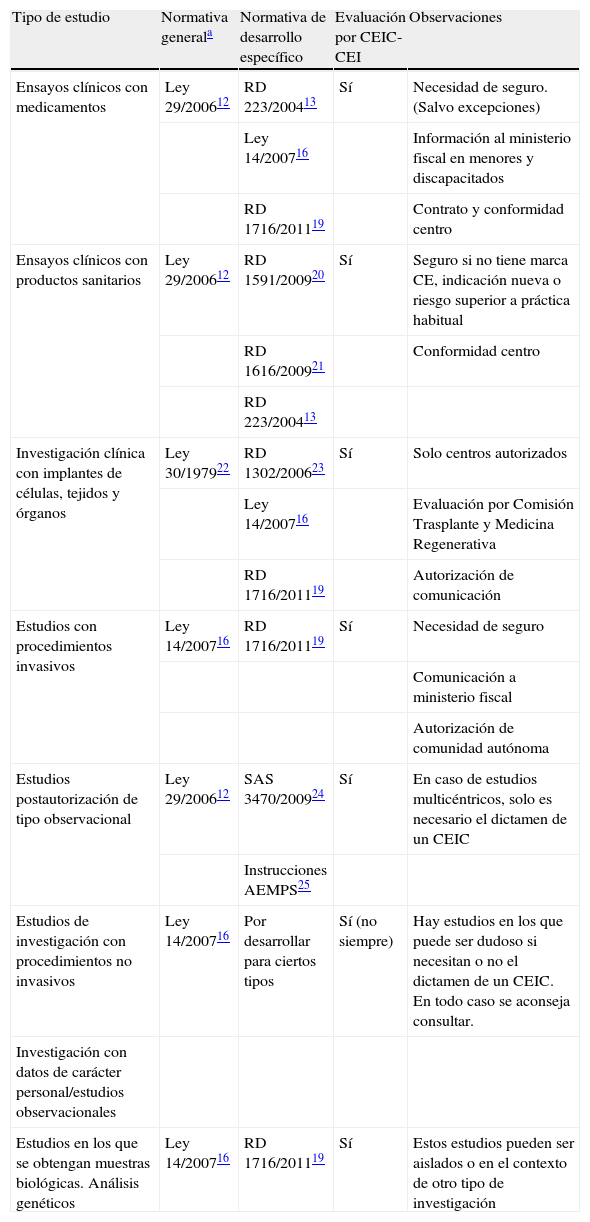
En la tabla 1 se relaciona el tipo de investigación descrita con la normativa correspondiente18.
Síntesis de normativa legal aplicable a los distintos tipos de investigación clínica
| Tipo de estudio | Normativa generala | Normativa de desarrollo específico | Evaluación por CEIC-CEI | Observaciones |
| Ensayos clínicos con medicamentos | Ley 29/200612 | RD 223/200413 | Sí | Necesidad de seguro. (Salvo excepciones) |
| Ley 14/200716 | Información al ministerio fiscal en menores y discapacitados | |||
| RD 1716/201119 | Contrato y conformidad centro | |||
| Ensayos clínicos con productos sanitarios | Ley 29/200612 | RD 1591/200920 | Sí | Seguro si no tiene marca CE, indicación nueva o riesgo superior a práctica habitual |
| RD 1616/200921 | Conformidad centro | |||
| RD 223/200413 | ||||
| Investigación clínica con implantes de células, tejidos y órganos | Ley 30/197922 | RD 1302/200623 | Sí | Solo centros autorizados |
| Ley 14/200716 | Evaluación por Comisión Trasplante y Medicina Regenerativa | |||
| RD 1716/201119 | Autorización de comunicación | |||
| Estudios con procedimientos invasivos | Ley 14/200716 | RD 1716/201119 | Sí | Necesidad de seguro |
| Comunicación a ministerio fiscal | ||||
| Autorización de comunidad autónoma | ||||
| Estudios postautorización de tipo observacional | Ley 29/200612 | SAS 3470/200924 | Sí | En caso de estudios multicéntricos, solo es necesario el dictamen de un CEIC |
| Instrucciones AEMPS25 | ||||
| Estudios de investigación con procedimientos no invasivos | Ley 14/200716 | Por desarrollar para ciertos tipos | Sí (no siempre) | Hay estudios en los que puede ser dudoso si necesitan o no el dictamen de un CEIC. En todo caso se aconseja consultar. |
| Investigación con datos de carácter personal/estudios observacionales | ||||
| Estudios en los que se obtengan muestras biológicas. Análisis genéticos | Ley 14/200716 | RD 1716/201119 | Sí | Estos estudios pueden ser aislados o en el contexto de otro tipo de investigación |
AEMPS: Agencia Española del Medicamento y Productos Sanitarios; CE: Comité Ético; CEIC-CEI: Comité Ético de Investigación Clínica-Comité de Ética de la Investigación.
Con objeto de que la investigación se desarrolle correctamente, respetando los derechos de los investigadores a la investigación a la vez que se garantizan los derechos fundamentales de los participantes, se han creado los Comités de Ética relacionados con la investigación. La nomenclatura de los mismos puede variar dependiendo de los distintos estados.
En España, tenemos los Comites Éticos de Investigación Clínica (CEIC), creados para la evaluación de EC y los Comités de Ética de la Investigación (CEI), que serían los encargados de evaluar otros aspectos de la IC, con excepción de los EC con medicamentos o productos sanitarios. En espera del desarrollo de la normativa que regule los CEI, son los CEIC los que en la práctica se encargan de la evaluación de todo tipo de IC, tal y como precisa la disposición adicional tercera de la Ley 14/2007 de Investigación Biomédica16.
Los CEIC son los órganos independientes, de composición multidisciplinar, constituidos por profesionales sanitarios y no sanitarios, encargados de velar por la protección de los derechos, seguridad y bienestar de los sujetos que participen en la investigación biomédica y además dar garantía pública de ello. Les corresponde evaluar los aspectos metodológicos, éticos y legales de los distintos tipos de proyectos de investigación que les sean remitidos.
Los comités que revisen propuestas de investigación pediátrica, sea del tipo que sea, deben contar con expertos en Pediatría que conozcan las necesidades especiales médicas, psicológicas y evolutivas de los niños. En la práctica, se requiere que al menos un miembro del comité sea un pediatra. En ocasiones, será necesaria la ayuda de expertos en ciertas especialidades pediátricas debido a la complejidad de los temas, como neonatología o cuidados intensivos.
Estos comités evalúan fundamentalmente:
- 1.
Aspectos relacionados con el principio de no maleficencia y justicia:
- –
Utilidad social de la investigación.
- –
La pertinencia y el diseño del proyecto.
- –
Aspectos metodológicos; un mal diseño del proyecto nunca será éticamente aceptable.
- –
Evaluación de los riesgos y beneficios previsibles para los sujetos participantes.
- –
Idoneidad del equipo investigador, colaboradores e instalaciones.
- –
Equiponderación clínica.
- –
Selección equitativa de la muestra.
- –
Previsión de compensación por daños (existencia de póliza de seguros).
- –
- –
- 2.
Aspectos relacionados con los principios de autonomía y de beneficencia:
- –
Información adecuada y obtención del consentimiento informado.
- –
Decisiones de sustitución por parte de los padres o de los representantes legales.
- –
Valoración subjetiva de la relación riesgo-beneficio desde la perspectiva del participante.
- –
Valoración de los límites de privacidad y confidencialidad.
- –
Expresión de las preferencias del participante.
- –
El plan previsto para el reclutamiento de los participantes.
- –
Las previsiones de remuneración para los investigadores.
- –
Las previsiones de compensación, si las hubiera, para los sujetos participantes.
- –
Otra de las funciones previstas de los comités es el seguimiento de los proyectos de investigación hasta la finalización de los mismos y el informe de los resultados. Específicamente, en los EC con medicamentos y productos sanitarios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.