





Puntos clave
- •
La muerte súbita de causa cardiaca en la población pediátrica es afortunadamente poco frecuente.
- •
Dos tercios de las enfermedades cardiacas que pueden producir muerte súbita en la población pediátrica tienen un patrón reconocible en el electrocardiograma (ECG).
- •
Los sustratos más habituales de la muerte súbita cardiaca son:
- –
La hipertrofia ventricular izquierda (ondas Q patológicas y alteraciones del ST y de la onda T en derivaciones izquierdas).
- –
La prolongación de la repolarización (QTc > 440–470ms en varones y > 460–480en mujeres).
- –
Las enfermedades arritmogénicas del ventrículo derecho (ensanchamiento del QRS, ascenso del ST y ondas T negativas en precordiales derechas).
- –
- –
Enfermedades potencialmente malignas pueden presentar en determinados momentos de la evolución un ECG normal.
La mayoría de los episodios de muerte súbita (MS) en niños, adolescentes y deportistas tiene una causa cardiaca1. La incidencia de MS en la población general oscila entre 50–60 casos/100.000 habitantes/año, de los cuales los niños y jóvenes representan una pequeña proporción2. En un reciente estudio poblacional de MS se registraron 33 casos en la edad pediátrica (0–18 años) en un período de 3 años, lo que supuso una incidencia de 1,7/100.000 habitantes/año. De estos 33 casos, 25 correspondieron a menores de un año; de estos 25 casos, 23 fueron inexplicados y 2 como consecuencia de cardiopatía congénita no diagnosticada3.
La MS de un niño o joven es un acontecimiento tan sumamente trágico para la familia y para la sociedad que ha motivado el llamamiento a realizar programas de detección precoz de las enfermedades que pueden ser sustrato de MS en distintas etapas de la vida o en determinados subgrupos de niños4. El electrocardiograma (ECG) es una herramienta de gran utilidad en el diagnóstico de la mayoría estas enfermedades. Sin embargo, el debate sobre el cribado cardiovascular en niños y jóvenes continúa sin resolverse y hasta el momento actual no existe suficiente evidencia científica para recomendar la realización de ECG a toda la población pediátrica5–7.
Causas de muerte súbita cardiaca en la edad pediátricaLas causas de MS cardiaca en niños y jóvenes se pueden agrupar en: a) cardiopatías estructurales, y b) trastornos eléctricos (tabla 1)8.
Causas de muerte súbita cardiaca en niños y adolescentes.
| Cardiopatías estructurales |
| Miocardiopatía hipertrófica |
| Anomalías coronarias |
| Miocardiopatía/displasia arritmogénica de ventrículo derecho |
| Miocardiopatía dilatada |
| Miocarditis |
| Estenosis aórtica grave |
| Dilatación de aorta ascendente |
| Trastornos eléctricos |
| Síndrome de Wolf-Parkinson-White |
| Canalopatías |
| Síndrome de QT largo |
| Síndrome de Brugada |
| Taquicardia ventricular polimórfica catecolaminérgica |
| Síndrome de QT corto |
En rojo aparecen las entidades que pueden ser detectadas mediante electrocardiograma.
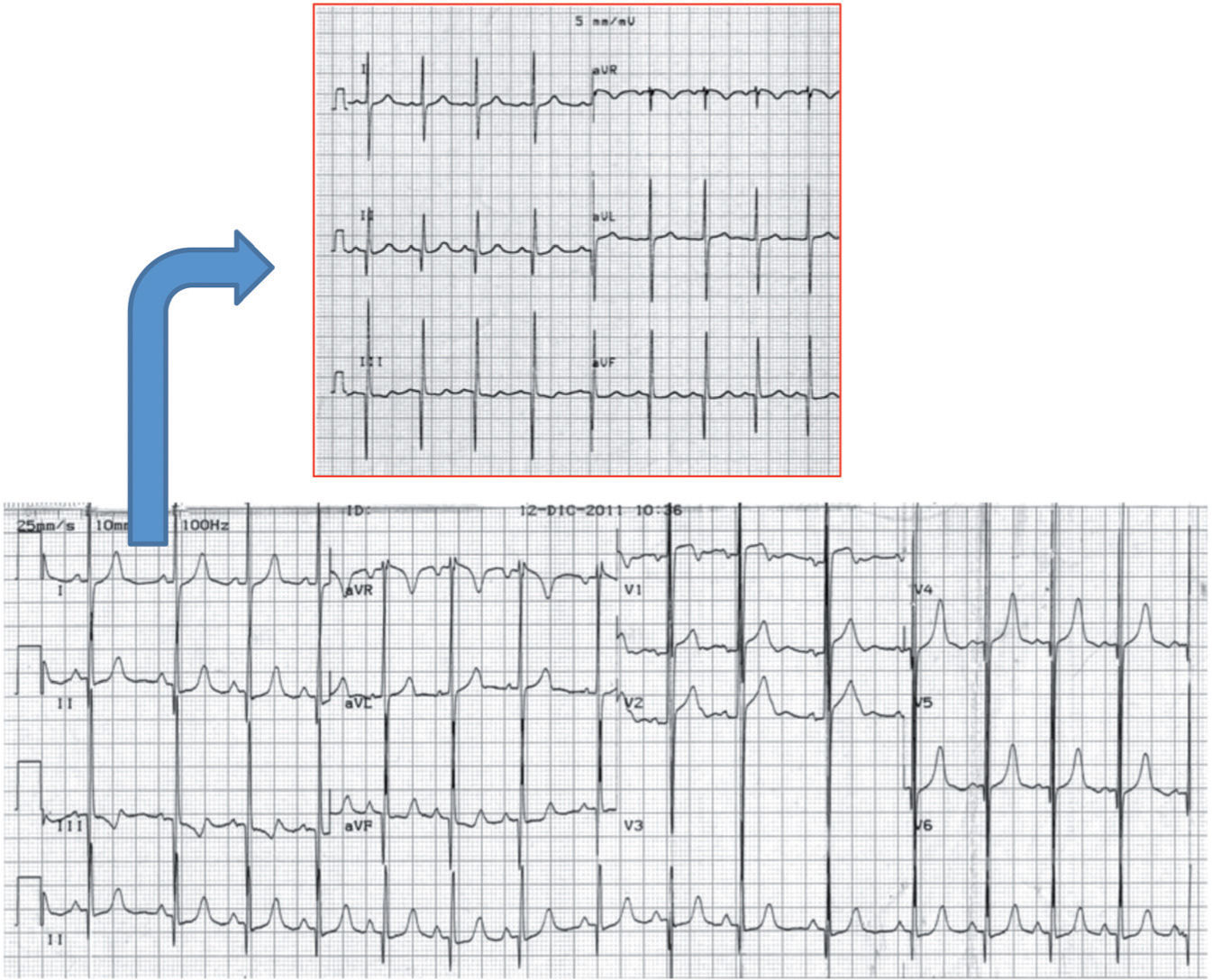
Es la principal causa de MS en deportistas menores de 35 años en las series americanas (aproximadamente un tercio de los casos)9. Se calcula una prevalencia en la población general de 1/500, aunque en la población pediátrica la prevalencia probablemente es menor, en torno a 1/15.000. La incidencia anual de MS en poblaciones no seleccionadas es alrededor del 1%10. En la miocardiopatía hipertrófica (MH), el ECG es anormal en el 95% de los casos11. La presencia aislada de criterios de voltaje de hipertrofia ventricular izquierda (HVI) es un hallazgo muy frecuente en niños sanos y en deportistas. Sin embargo, la HVI patológica (fig. 1) presenta además criterios no dependientes de voltaje: ondas Q patológicas en derivaciones inferiores y/o laterales, cambios muy prominentes en el segmento ST y en la onda T.
 y ascenso del ST en precordiales anteriores. Figura adjunta: detalle de las derivaciones del plano frontal del mismo paciente en registro realizado con calibración 5mm/mV para apreciar con detalle las ondas Q.")
Registro electrocardiográfico de niño de 7 años con diagnóstico de miocardiopatía hipertrófica no obstructiva familiar. Presenta aumento de voltaje de los complejos QRS, ondas Q patológicas en derivaciones inferiores (II, III y aVF) y ascenso del ST en precordiales anteriores. Figura adjunta: detalle de las derivaciones del plano frontal del mismo paciente en registro realizado con calibración 5mm/mV para apreciar con detalle las ondas Q.
Las anomalías coronarias que con mayor frecuencia pueden dar lugar a MS en la edad pediátrica son aquellas en las que existe un origen anómalo de una arteria coronaria12.
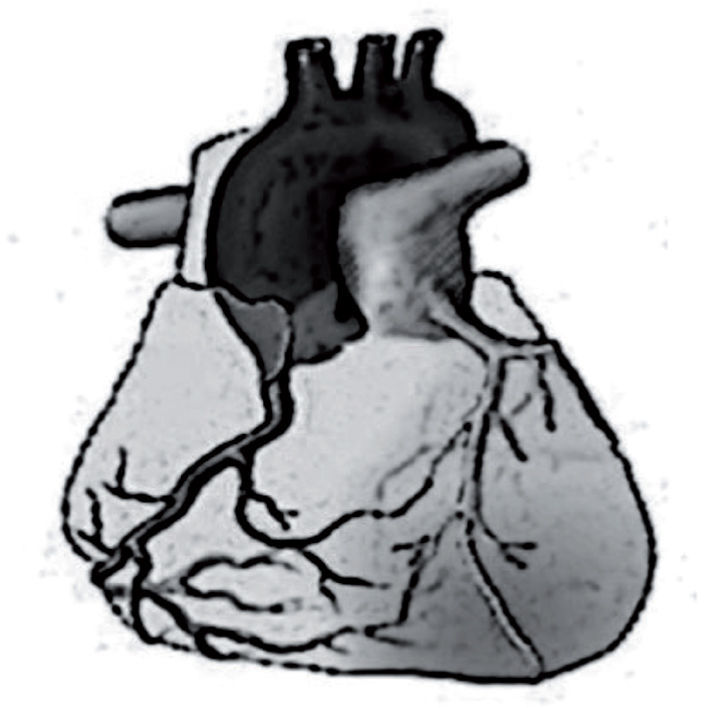
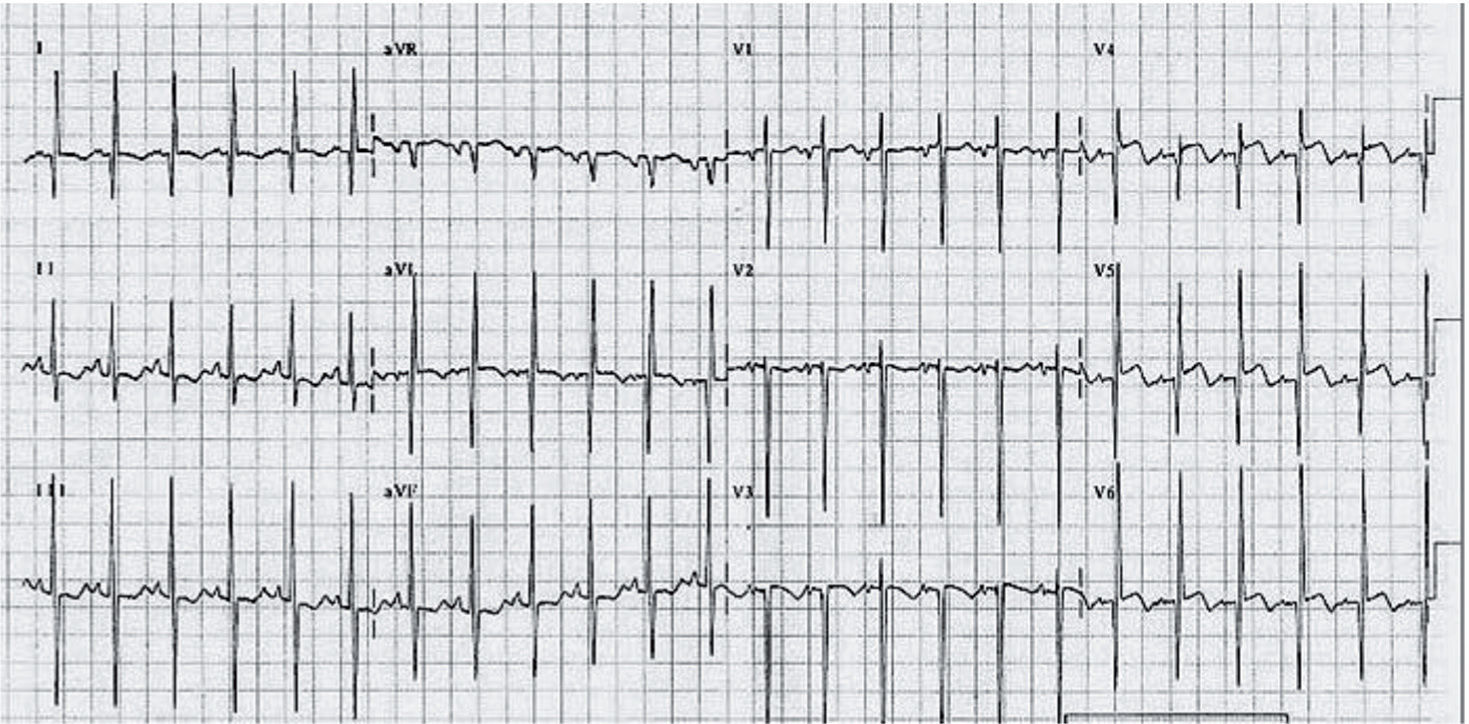
El origen anómalo de la arteria coronaria izquierda (ACI) en la arteria pulmonar representa entre el 0,25 y el 0,5% de las cardiopatías congénitas13. La clínica aparece característicamente a partir del mes de vida, en relación con la caída fisiológica de presiones pulmonares y la inversión del flujo de la ACI anómala en dirección al tronco pulmonar (fig. 2), y es de isquemia coronaria, habitualmente difícil de identificar en lactantes. Hasta el 40% de los casos puede presentarse en forma de MS. El corazón está dilatado y presenta fibrosis y adelgazamiento de la pared anterolateral infartada del ventrículo izquierdo. El ECG muestra ondas Q anchas y profundas, así como elevación del ST en las derivaciones que recogen el área infartada (I, aVL, V4–V6). El hallazgo de ondas Q profundas (> 3mm) con onda T invertida en aVL es específico de esta entidad (fig. 3).

Origen anómalo de la arteria coronaria izquierda en la arteria pulmonar. Las resistencias pulmonares elevadas características del período fetal permiten la perfusión anterógrada de la coronaria izquierda a partir de la arteria pulmonar. Después del parto, las resistencias pulmonares empiezan a bajar y sobre los 2 meses de vida ya alcanzan los niveles del adulto. En esta situación hemodinámica, el flujo de la coronaria anómala se invierte y se dirige hacia el tronco pulmonar robando el flujo procedente de las colaterales y produciendo isquemia de la cara anterolateral del ventrículo izquierdo.

Registro electrocardiográfico de lactante mujer de 4 meses con diagnóstico de origen anómalo de la arteria coronaria izquierda en la arteria pulmonar con comienzo en forma de shock cardiogénico. Patrón de infarto anterolateral con ondas Q profundas en I, aVL y V4–V6 y ascenso del ST en V4–V6. Obsérvese el patrón característico de esta entidad: onda Q patológica en aVL con inversión de la onda T.
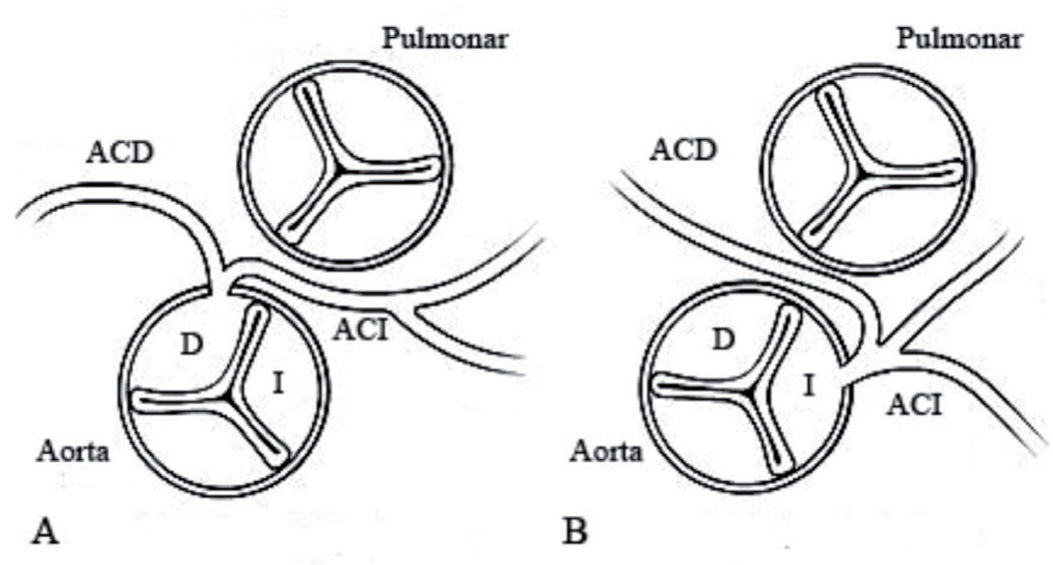
El origen anómalo de una arteria coronaria en el seno del Valsalva opuesto (ACI en seno de Valsalva derecho o arteria coronaria derecha en seno de Valsalva izquierdo) (fig. 4) tiene una prevalencia muy baja14. Característicamente, producen MS en niños mayores y jóvenes en relación con ejercicio físico intenso. Sin embargo, el ECG en reposo generalmente no muestra alteraciones15.
 Origen anómalo de la arteria coronaria izquierda (ACI) en el seno de Valsalva derecho. B) Origen anómalo de la arteria coronaria derecha (ACD) en el seno de Valsalva izquierdo. El recorrido obligado entre las dos grandes arterias junto con anomalías frecuentes asociadas como la forma del ostium (en forma de hendidura), el origen en ángulo agudo, pueden determinar alteraciones de la perfusión coronaria que se ponen de manifiesto sólo en momentos de máximo esfuerzo.")
A) Origen anómalo de la arteria coronaria izquierda (ACI) en el seno de Valsalva derecho. B) Origen anómalo de la arteria coronaria derecha (ACD) en el seno de Valsalva izquierdo. El recorrido obligado entre las dos grandes arterias junto con anomalías frecuentes asociadas como la forma del ostium (en forma de hendidura), el origen en ángulo agudo, pueden determinar alteraciones de la perfusión coronaria que se ponen de manifiesto sólo en momentos de máximo esfuerzo.
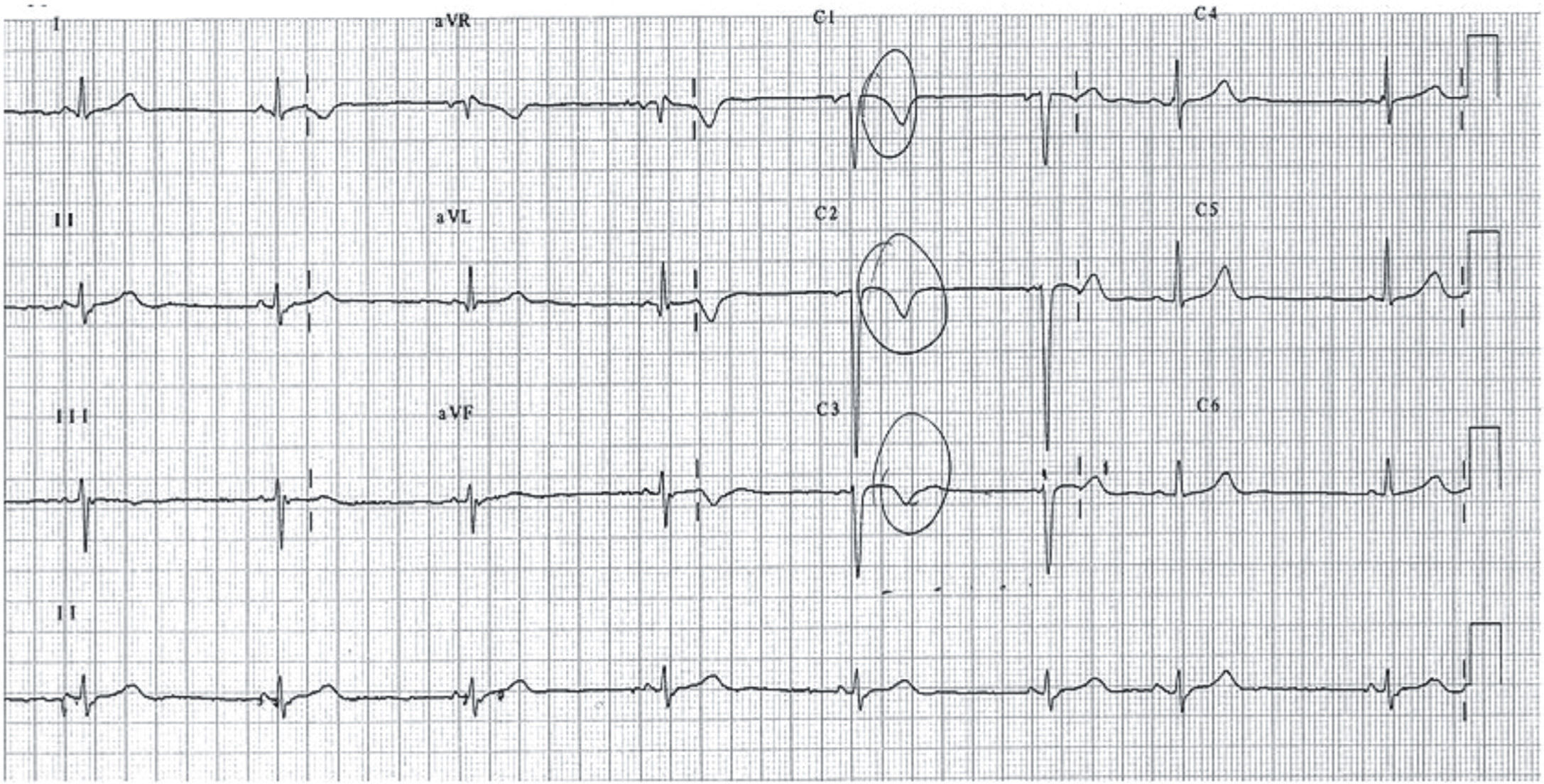
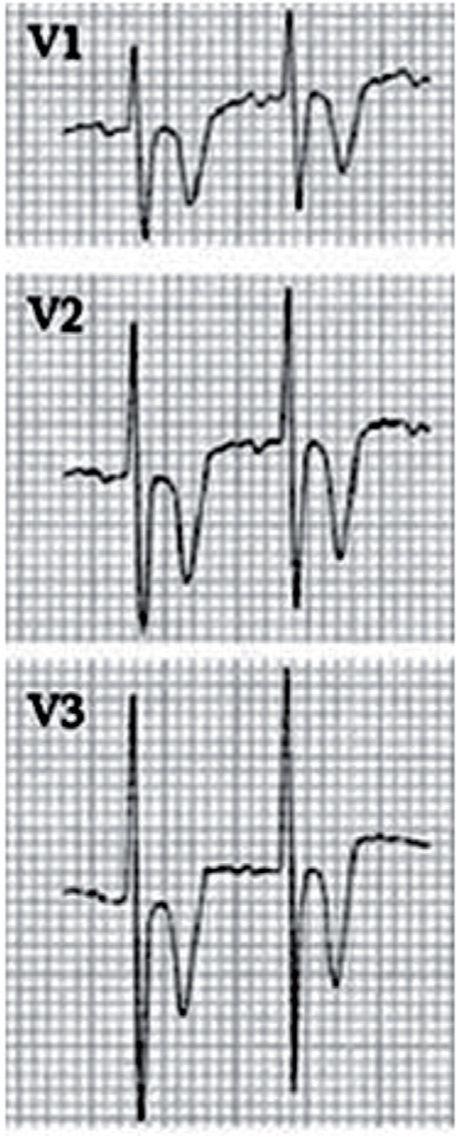
La displasia arritmogénica de ventrículo derecho (DAVD) se caracteriza por el reemplazamiento del miocardio del ventrículo derecho (VD) por tejido fibroadiposo. La prevalencia estimada para la población general varía entre 1/2.000 y 1/5.000, con una relación varón/mujer de 3:1. Es el trastorno estructural más frecuentemente asociado a MS en deportistas < 30 años en España16. Se trata de una enfermedad hereditaria que se transmite fundamentalmente de forma autosómica dominante, con reducida penetrancia y expresión variable y se han descrito 12 genes ligados a la DAVD que codifican distintos componentes de las uniones intercelulares17. Es una enfermedad poco frecuente en la edad pediátrica, excepcional antes en la primera década de la vida. La presentación típica es en forma de arritmias ventriculares sintomáticas con origen en el VD (desde extrasístoles ventriculares aisladas a rachas sostenidas de taquicardia ventricular con morfología de bloqueo de rama izquierda), generalmente desencadenadas por el esfuerzo. Los hallazgos ECG característicos incluyen la presencia de ondas T negativas en V1–V3 (85% de pacientes), ensanchamiento del QRS limitado a precordiales derechas sin imagen de bloqueo de rama derecha (QRS > 110ms en V1–V3; 64%) y ondas épsilon (muesca o espícula terminal en el complejo QRS) en precordiales derechas (33%) (figs. 5 y 6).
. El patrón juvenil normal de repolarización hace referencia a la presencia de ondas T negativas en precordiales anteriores hasta aproximadamente los 10 años. A partir de esta edad, la onda T se debe hacer positiva en la cara anterior (progresivamente desde V3 a V1), aunque en el adulto joven la onda T puede seguir siendo negativa en V1. Por tanto, en este adolescente sería normal encontrar una onda T negativa en V1, pero no en V2 y mucho menos en V3. Con el antecedente de síncope, es obligado descartar DAVD.")
Registro electrocardiográfico de varón de 14 años estudiado por síncope y diagnosticado de displasia arritmogénica de ventrículo derecho (DAVD). El patrón juvenil normal de repolarización hace referencia a la presencia de ondas T negativas en precordiales anteriores hasta aproximadamente los 10 años. A partir de esta edad, la onda T se debe hacer positiva en la cara anterior (progresivamente desde V3 a V1), aunque en el adulto joven la onda T puede seguir siendo negativa en V1. Por tanto, en este adolescente sería normal encontrar una onda T negativa en V1, pero no en V2 y mucho menos en V3. Con el antecedente de síncope, es obligado descartar DAVD.
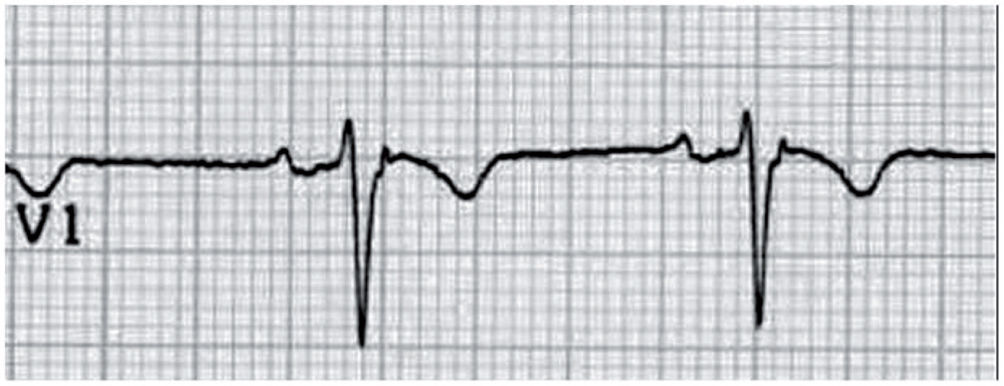
. Detalle que demuestra el ensanchamiento del QRS en V1 sin imagen de bloqueo de rama derecha y una muesca o espícula terminal, la onda épsilon, que corresponde a potenciales eléctricos retrasados de pequeña amplitud originados en las áreas de tejido sano rodeadas de tejido fibroadiposo.")
Displasia arritmogénica de ventrículo derecho (DAVD). Detalle que demuestra el ensanchamiento del QRS en V1 sin imagen de bloqueo de rama derecha y una muesca o espícula terminal, la onda épsilon, que corresponde a potenciales eléctricos retrasados de pequeña amplitud originados en las áreas de tejido sano rodeadas de tejido fibroadiposo.
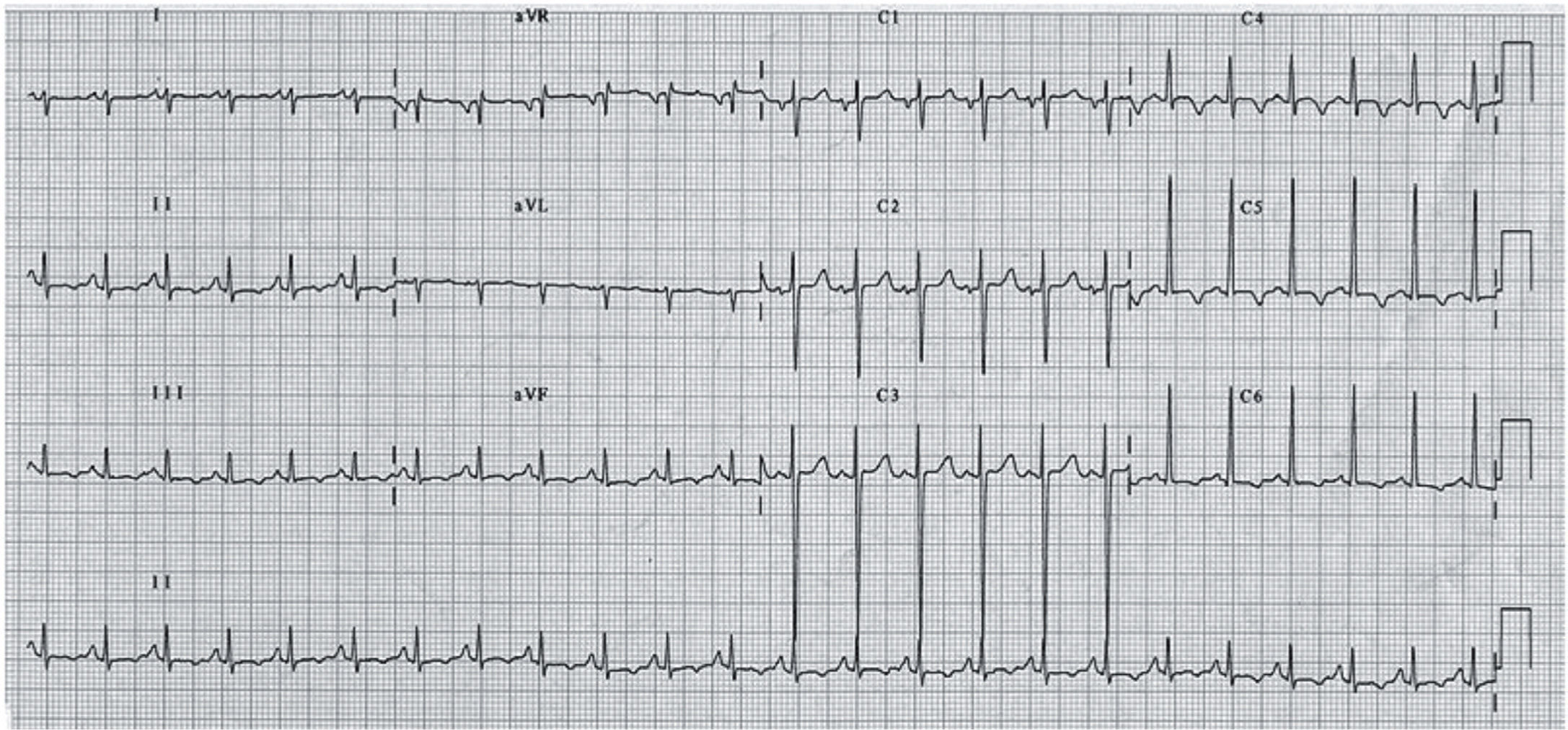
La miocardiopatía dilatada (MD) es la forma más frecuente de miocardiopatía y en la edad pediátrica afecta especialmente a pacientes menores de un año. Habitualmente solo se consigue identificar la etiología en un tercio de los pacientes18. La presentación suele ser en forma de insuficiencia cardiaca en más del 70%, pero también puede ser diagnosticada casualmente en base a un ECG patológico. En la fase presintomática, el riesgo de MS es bajo, pero significativamente superior al de la población general. La MD da lugar a una serie de cambios ECG no específicos en forma de alteraciones del ST y de la onda T (fig. 7).
Miocarditis
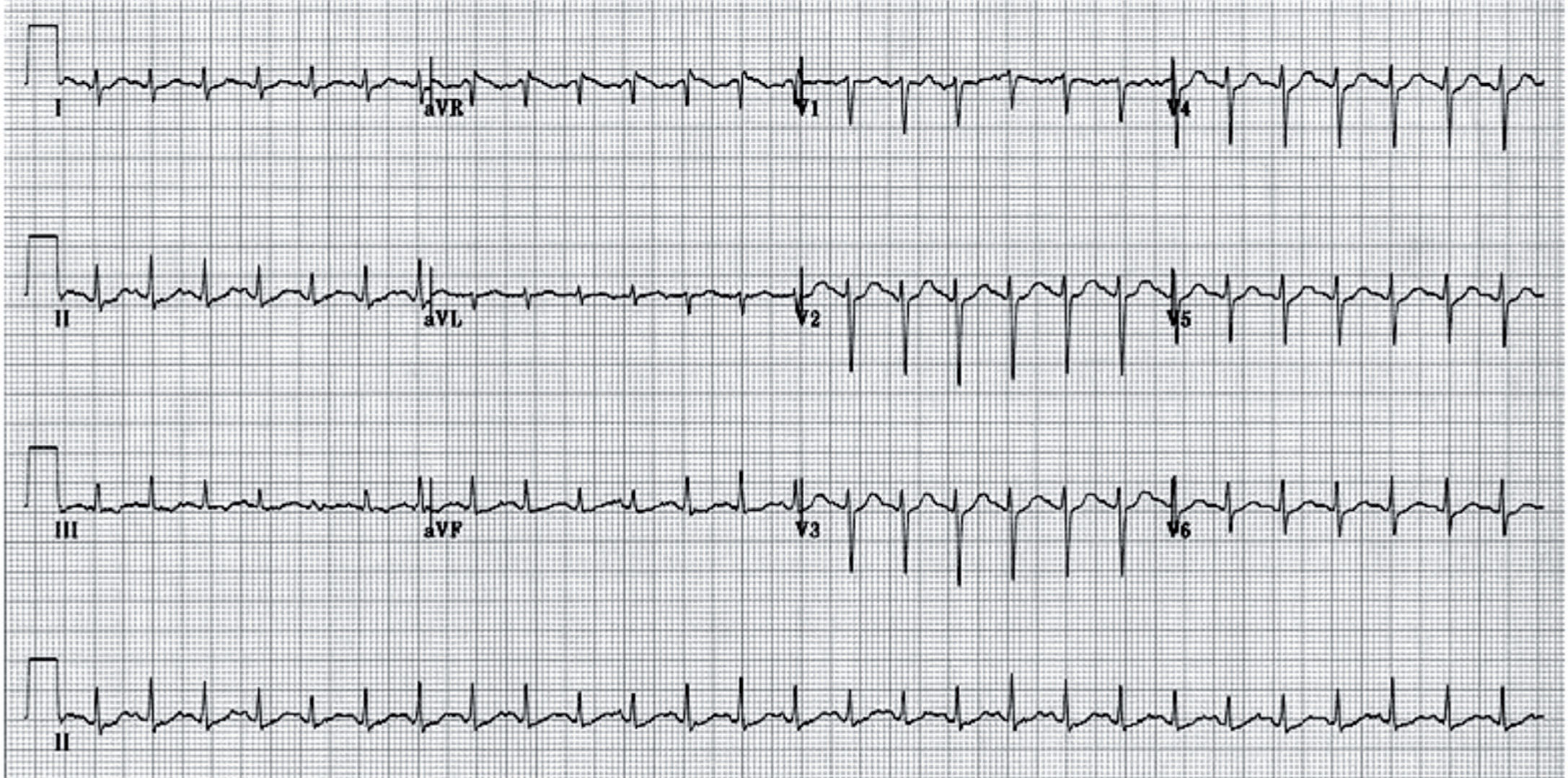
La miocarditis es una enfermedad habitualmente leve o subclínica pero puede cursar de forma fulminante o como sustrato de MS, especialmente en lactantes19. En el ECG se puede encontrar cualquier combinación de los siguientes hallazgos (fig. 8): taquicardia sinusal, complejos QRS de bajo voltaje, cambios en el segmento ST y en la onda T, prolongación del PR, prolongación del intervalo QTc.
Estenosis aórtica grave constituyen los hallazgos más frecuentes.")
La estenosis aórtica valvular y/o supravalvular grave, a pesar de ser fácilmente reconocible desde el punto de vista clínico, sigue siendo causa de MS en relación con esfuerzo16. El descenso del ST y la inversión de la onda T en derivaciones izquierdas es un marcador ECG característico de hipertrofia ventricular izquierda grave que traduce isquemia latente y alto riesgo de MS (fig. 9).
Síndrome de Wolf-Parkinson-White con ondas T negativas (aVL, V5 y V6) traduce hipertrofia ventricular izquierda grave con isquemia latente.")
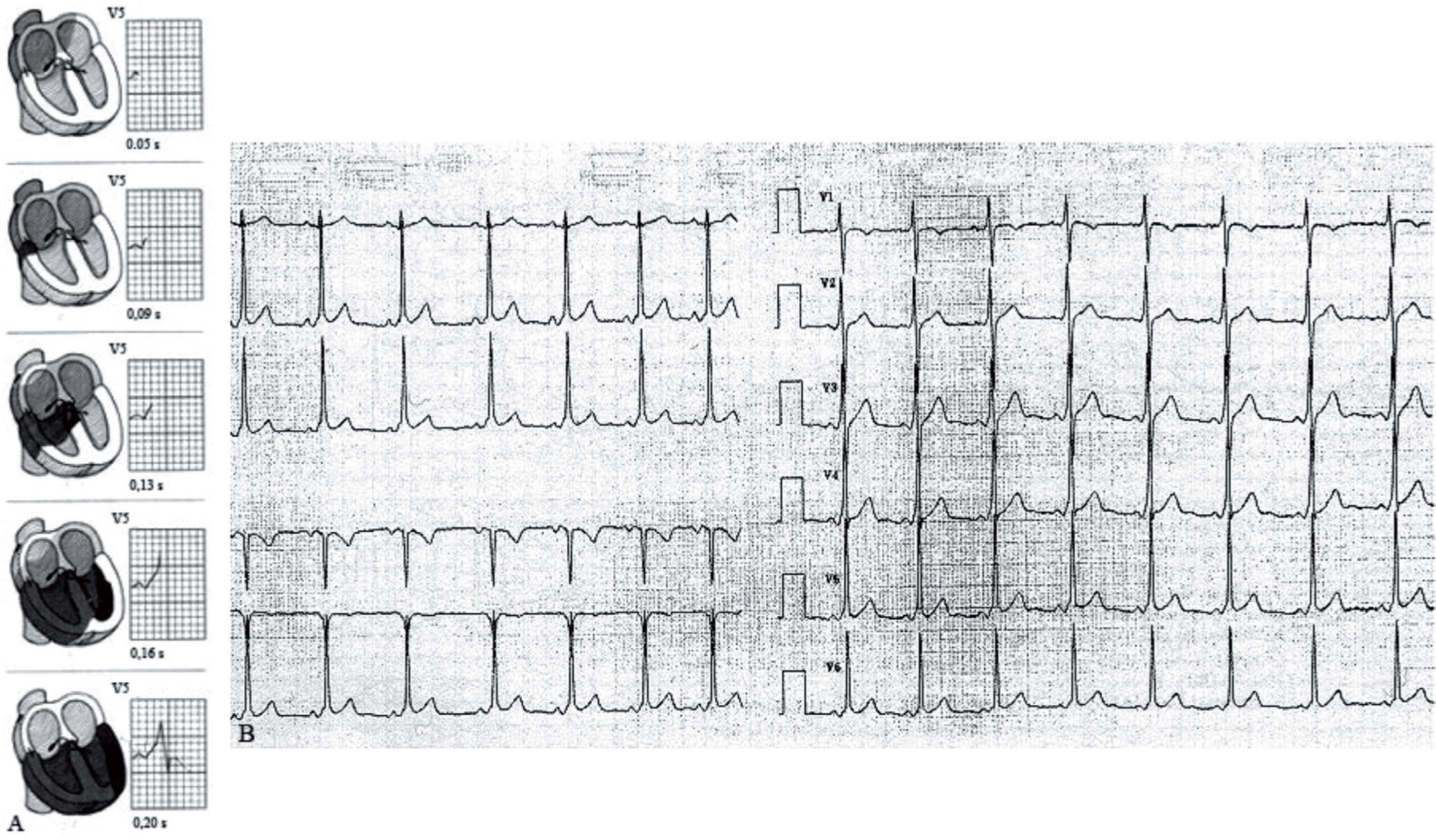
La preexcitación tipo Wolf-Parkinson-White (WPW) se produce por la presencia de una vía accesoria que conecta aurículas con ventrículos y que permite que una parte del miocardio ventricular se despolarice inmediatamente después de la despolarización auricular (preexcitación), dando lugar a una onda lenta de activación ventricular (onda delta) que configura un intervalo PR corto y un ensanchamiento del QRS (fig. 10). El síndrome de WPW (preexcitación más episodios de taquicardia supraventricular) se completa cuando la vía accesoria tiene además capacidad para conducir en sentido retrógrado (reentrada desde ventrículo a aurícula).
 Preexcitación tipo Wolf-Parkinson-White. Después de despolarizarse las aurículas, el impulso alcanza la vía accesoria localizada en la pared lateral derecha y empieza a despolarizar de forma lenta el miocardio ventricular adyacente dando lugar a la onda delta. Después del retraso fisiológico de la conducción en el nodo auriculoventricular, el impulso se conduce de forma rápida por el sistema de conducción y despolariza el resto del miocardio. El QRS ancho resultante corresponde a la fusión de ambos frentes. B) Registro electrocardiográfico de varón de 9 años con preexcitación tipo Wolf-Parkinson-White. Presenta un PR corto con QRS ensanchado por la presencia de una onda delta.")
A) Preexcitación tipo Wolf-Parkinson-White. Después de despolarizarse las aurículas, el impulso alcanza la vía accesoria localizada en la pared lateral derecha y empieza a despolarizar de forma lenta el miocardio ventricular adyacente dando lugar a la onda delta. Después del retraso fisiológico de la conducción en el nodo auriculoventricular, el impulso se conduce de forma rápida por el sistema de conducción y despolariza el resto del miocardio. El QRS ancho resultante corresponde a la fusión de ambos frentes. B) Registro electrocardiográfico de varón de 9 años con preexcitación tipo Wolf-Parkinson-White. Presenta un PR corto con QRS ensanchado por la presencia de una onda delta.
Se estima un prevalencia de preexcitación en la población general de 2–4/1.000. La incidencia de WPW sintomático está en torno a 4/100.000/año, con un patrón bimodal: un pico en la infancia y otro entre los 20 y 30 años.
La presentación clínica habitual es en forma de episodios de taquicardia supraventricular por reentrada. Sin embargo, se describe un riesgo de MS asociada al WPW de 0,17% anual en pacientes asintomáticos, que parece ser ligeramente mayor en niños y adolescentes (hasta 0,3% anual)20. Se admite que el mecanismo de la MS en estos pacientes es conducción rápida a los ventrículos de un episodio de fibrilación auricular (FA) a través de una vía accesoria con período refractario corto. Aunque la FA es excepcional en la edad pediátrica, es conocido que la práctica de deporte incrementa el riesgo de desarrollar FA21, probablemente como consecuencia del incremento del tono vagal (acortamiento del período refractario auricular) y del desarrollo de cambios anatómicos a nivel auricular (aumento de tamaño de las aurículas, etc.), por lo que el hallazgo de preexcitación tipo WPW requiere una evaluación cardiológica dirigida a determinar el período refractario de la vía anómala. La desaparición de la preexcitación con frecuencias cardiacas superiores a 120–130lpm sugiere un período refractario largo y se ha considerado tradicionalmente un factor de buen pronóstico22.
Síndrome de QT largoEl síndrome de QT largo (STQL) congénito es la canalopatía responsable del mayor número de MS de causa arrítmica en niños y jóvenes. Se ha publicado una prevalencia de al menos 1/2.500 recién nacidos vivos23. Se trata de un trastorno genético heterogéneo que se hereda fundamentalmente de forma autosómica dominante, con penetrancia variable. Se han identificado cientos de mutaciones en los 13 genes que participan en la codificación de los canales iónicos, integrados por una unidad principal o subunidad alfa y una serie de proteínas auxiliares que lo regulan. Aproximadamente, el 75% de pacientes con diagnóstico clínico de SQTL presentan mutaciones en alguno de los 3 principales genes que codifican la subunidad alfa y constituyen los 3 tipos de SQTL más frecuentes: KCNQ1 (SQTL tipo 1; canal de potasio IKs; 35%); KCNH2 (SQTL tipo 2; canal de potasio IKr; 30%) y SCN5A (SQTL tipo 3; canal de sodio; 10%).
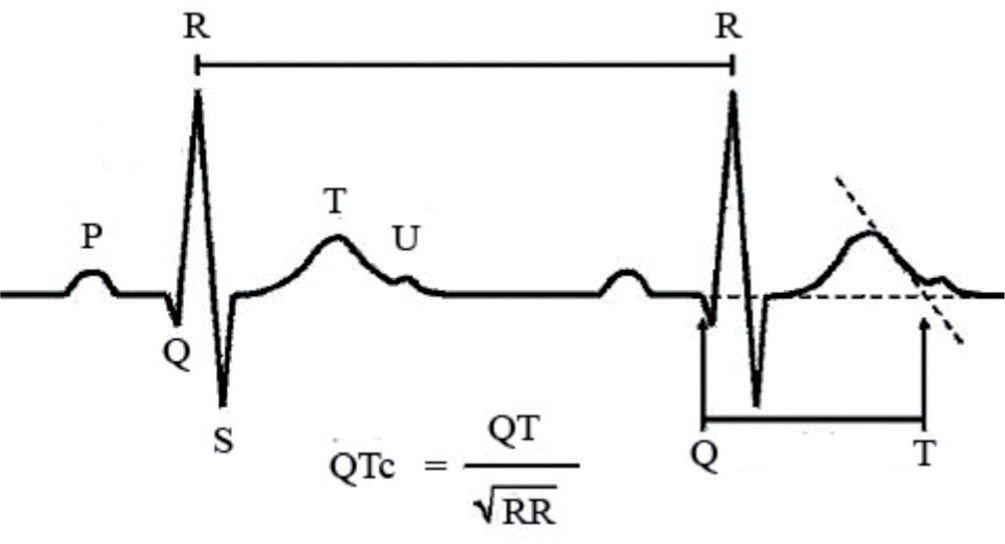
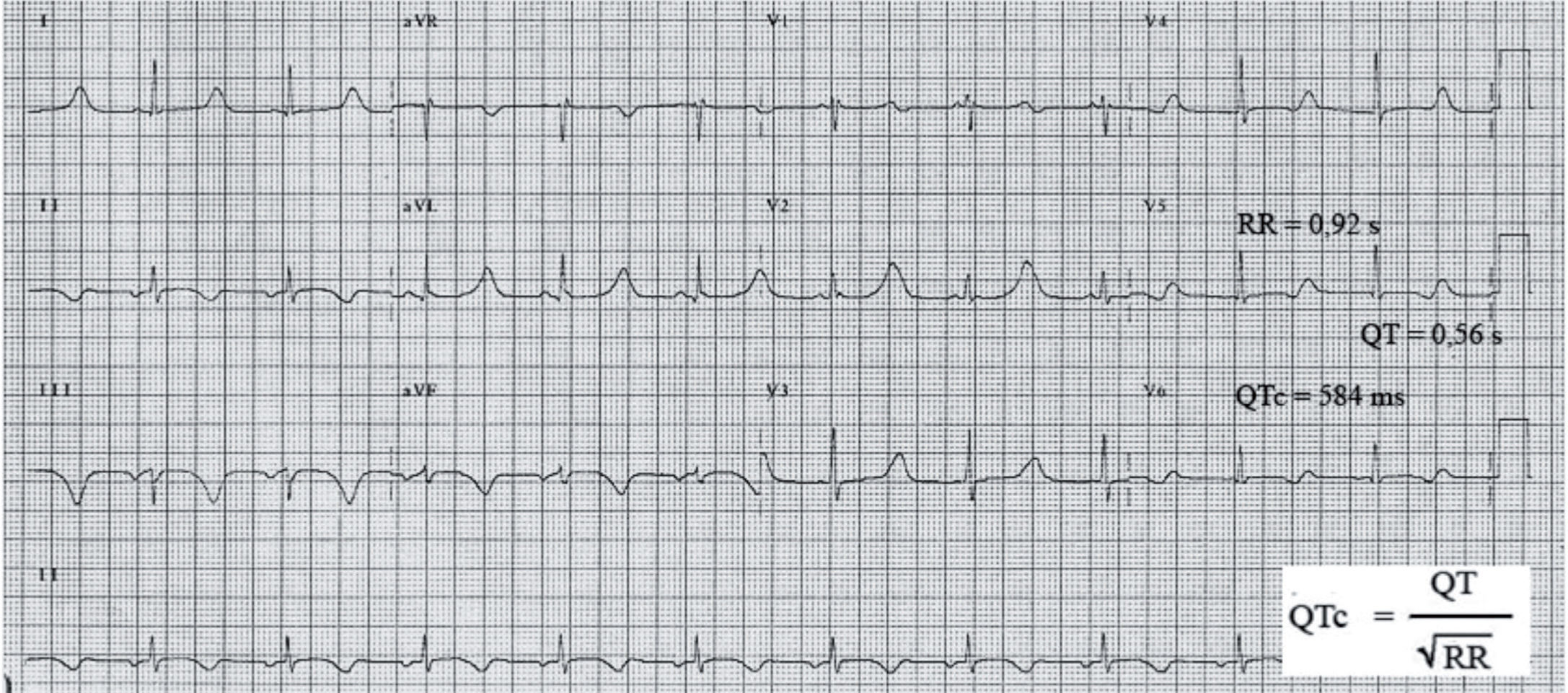
El SQTL ha dejado de ser una curiosidad médica y en el momento actual todos los clínicos deben estar familiarizados con la manera de medir correctamente el intervalo QT (figs. 11 y 12)24,25. Se considera anormal un QTc > 440ms en varones y > 460ms en mujeres. Sin embargo, el QT es un intervalo dinámico y puede existir cierto grado de solapamiento entre los valores de QTc de pacientes con SQTL y la población normal por lo que algunos autores han propuesto considerar QTc anormal > 470ms en varones y > 480 en mujeres26.

Medición del intervalo QTc: se utiliza la fórmula de Bazett para corregir la duración del intervalo de acuerdo con la frecuencia cardiaca. Debe medirse preferentemente en II y en V5 o en la derivación en que parezca ser más largo. Debe delimitarse el final de la onda T, con ayuda del método de la tangente como se muestra en la figura25, con respecto a la onda U. El QT medido de esta manera se divide entre la raíz cuadrada del intervalo RR precedente. Velocidad: 25mm/s; 1mm = 0,04s. De forma rápida: se debe sospechar QT largo cuando la onda T supera la mitad del intervalo RR o bien cuando se «monta» sobre la onda P del siguiente ciclo cardiaco.
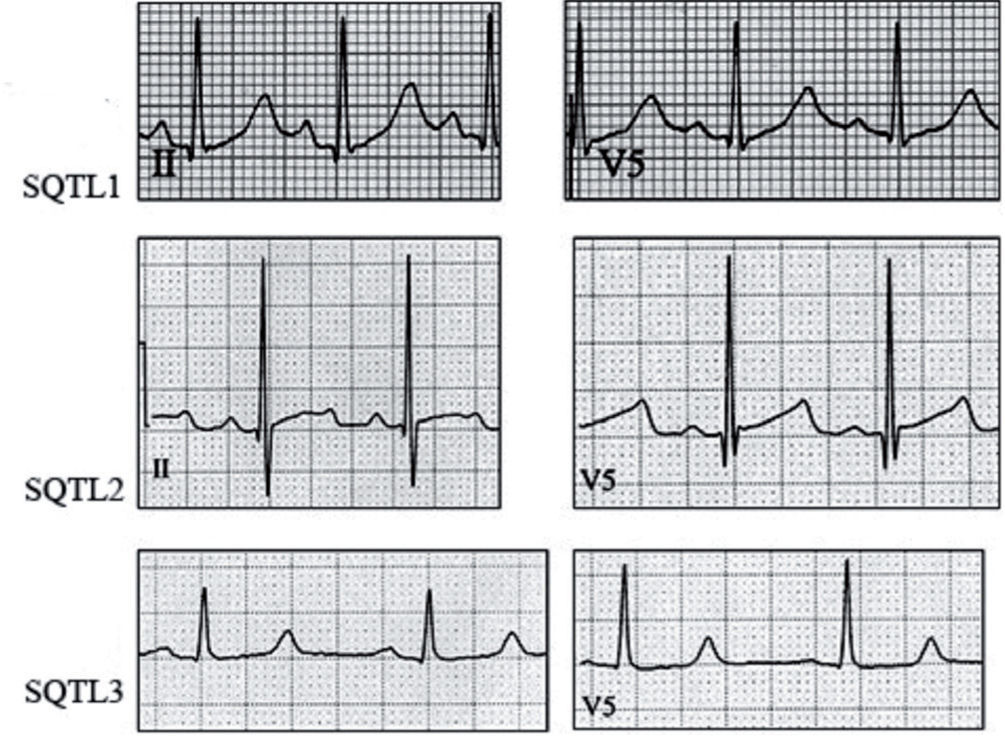
. Subtipos más frecuentes25. SQTL tipo 1: ondas T de base ancha y duración muy prolongada; desencadenantes: ejercicio y/o estímulos simpáticos; especialmente natación. SQTL tipo 2: ondas T de baja amplitud, con muescas y con apariencia asimétrica; desencadenantes: emociones, estímulos auditivos bruscos; menos frecuente, sueño y ejercicio. SQTL tipo 3: ondas T de aparición tardía, de amplitud normal, simétricas; desencadenantes: sueño, bradicardia.")
Síndrome de QT largo (SQTL). Subtipos más frecuentes25. SQTL tipo 1: ondas T de base ancha y duración muy prolongada; desencadenantes: ejercicio y/o estímulos simpáticos; especialmente natación. SQTL tipo 2: ondas T de baja amplitud, con muescas y con apariencia asimétrica; desencadenantes: emociones, estímulos auditivos bruscos; menos frecuente, sueño y ejercicio. SQTL tipo 3: ondas T de aparición tardía, de amplitud normal, simétricas; desencadenantes: sueño, bradicardia.
El ECG permite identificar clínicamente el subtipo de SQTL (fig. 13) y constituye una herramienta de gran valor en la estratificación del riesgo (pacientes con QTc > 500ms tienen un riesgo superior al 50% de síncope y/o MS).
. QT medido en V5: 14mm×0,04s=0,56s; RR precedente: 23mm×0,04s=0,92s; raíz cuadrada de 0,92=0,959; QTc=QT medido/raíz cuadrada del RR precedente = 0,56/0,959=0,584s=584ms.")
Registro electrocardiográfico de paciente varón con síndrome de QT largo y diagnóstico previo de epilepsia (1mm=0,04s). QT medido en V5: 14mm×0,04s=0,56s; RR precedente: 23mm×0,04s=0,92s; raíz cuadrada de 0,92=0,959; QTc=QT medido/raíz cuadrada del RR precedente = 0,56/0,959=0,584s=584ms.
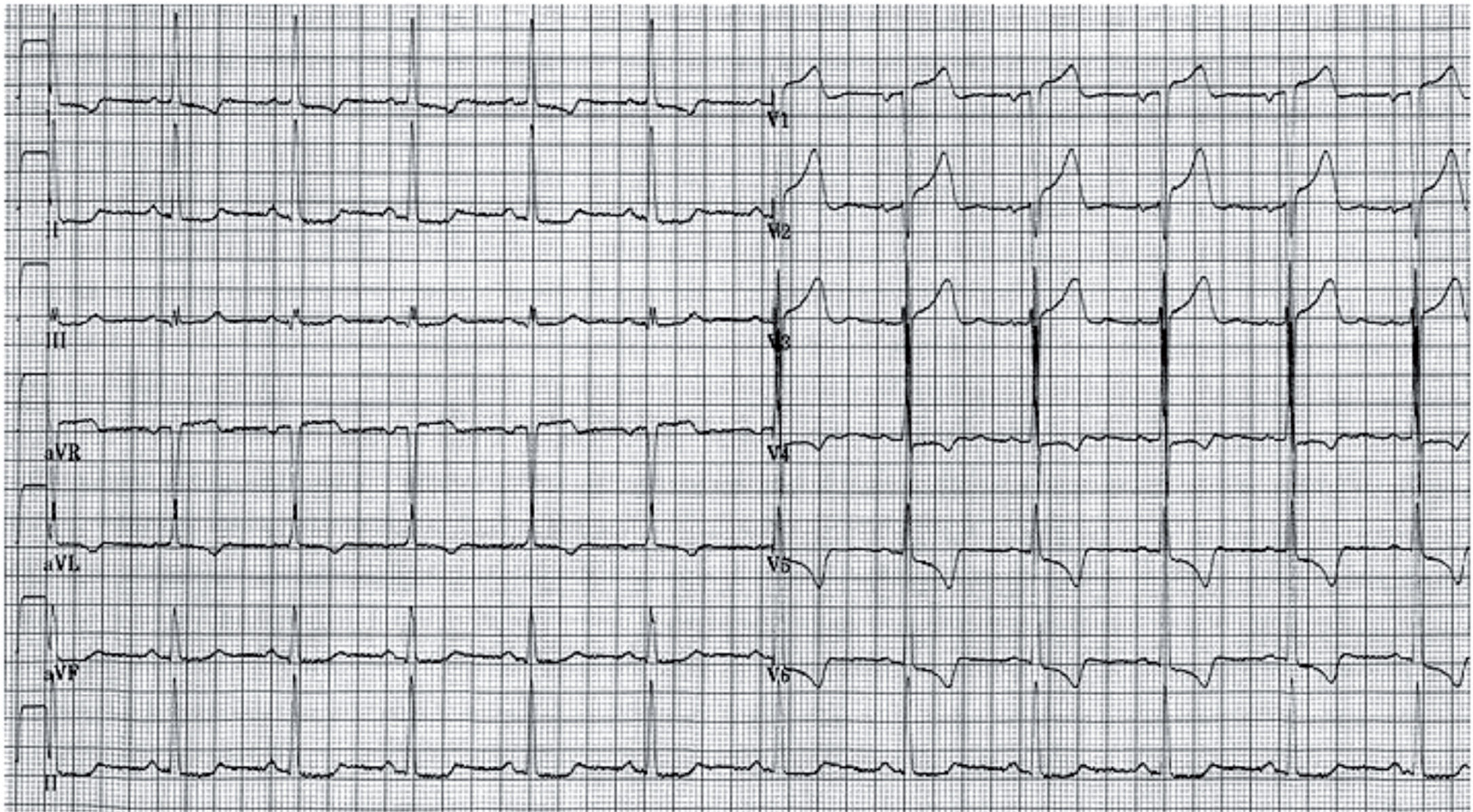
El síndrome de Brugada (SBr) es una canalopatía genéticamente heterogénea, con herencia autosómica dominante en aproximadamente el 50% de los casos y con variable penetrancia. Aunque recientemente se han descrito mutaciones en otros genes, el 20% de pacientes presenta mutaciones para el gen SCN5A que codifica para el canal de sodio. La prevalencia del SBr en la población occidental es probablemente baja. Sin embargo, en ciertas zonas del sudeste asiático, donde se conoce como síndrome de la MS nocturna inesperada, puede alcanzar una prevalencia de 1/2.000 y constituye la segunda causa de muerte en varones menores de 40 años27.
El diagnóstico del SBr se establece sobre la base del ECG. En el momento actual, se admite la existencia de 2 patrones28: tipo 1: elevación del ST ≥ 2mm, con inversión de la onda T en más de una derivación precordial derecha (fig. 14); tipo 2: elevación del ST ≥ 2mm en precordiales derechas con onda T positiva o isodifásica (fig. 15). Existen factores moduladores que pueden hacer que el ECG de un paciente con SBr muestre distintos patrones a lo largo del tiempo y que incluso pueda ser normal en algún momento. Es conocido que la temperatura es un modulador importante, lo que explica que la fiebre pueda desenmascarar formas silentes del SBr e incluso desencadenar arritmias ventriculares en el seno de un proceso febril29,30.
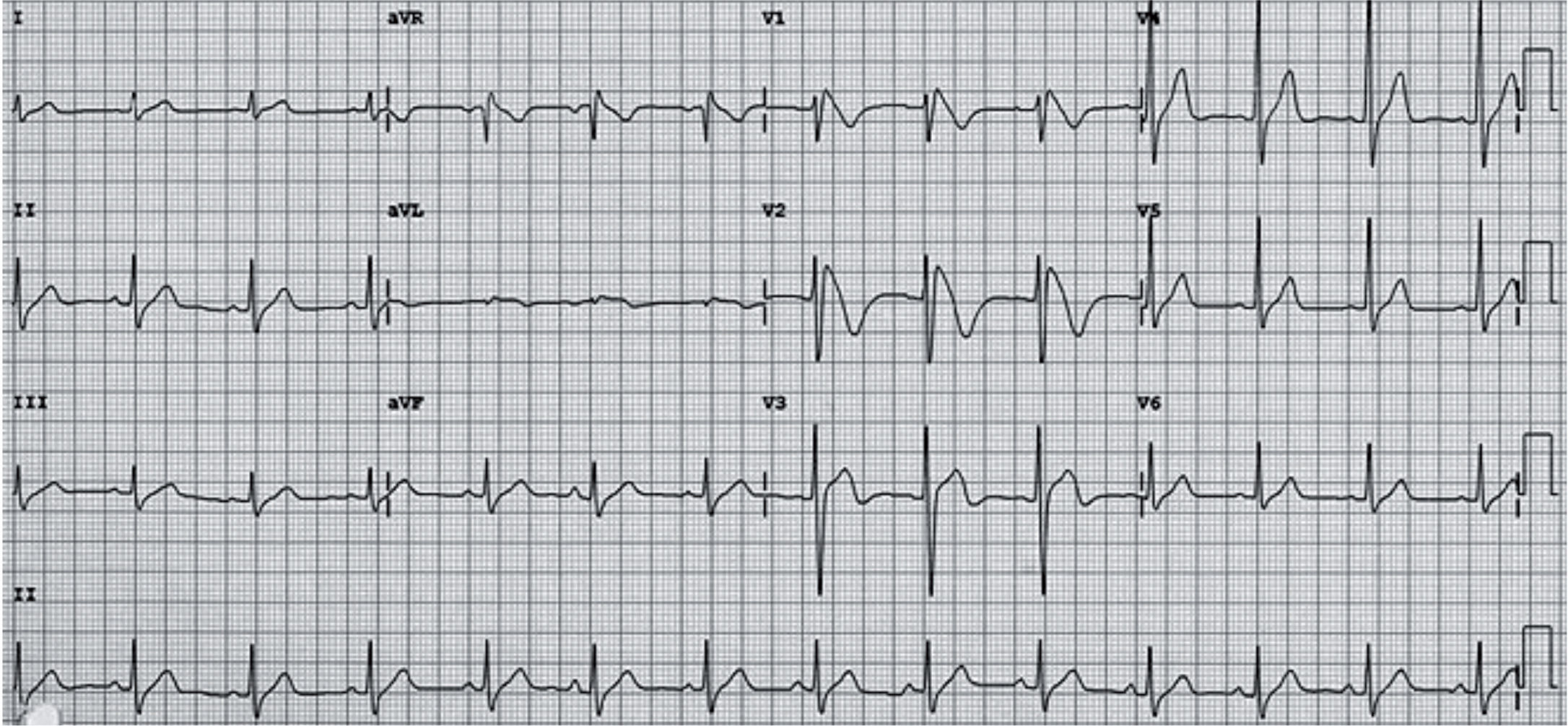
 de varón de 15 años que ha sufrido un episodio de muerte súbita abortada después de un ejercicio de resistencia y en el que se objetiva un patrón Brugada tipo 1. Se puede observar el ascenso del ST > 2mm en V1 y V2 (en forma de «aleta de tiburón»), así como inversión de la onda T en V1 y V2.")
Registro electrocardiográfico (ECG) de varón de 15 años que ha sufrido un episodio de muerte súbita abortada después de un ejercicio de resistencia y en el que se objetiva un patrón Brugada tipo 1. Se puede observar el ascenso del ST > 2mm en V1 y V2 (en forma de «aleta de tiburón»), así como inversión de la onda T en V1 y V2.
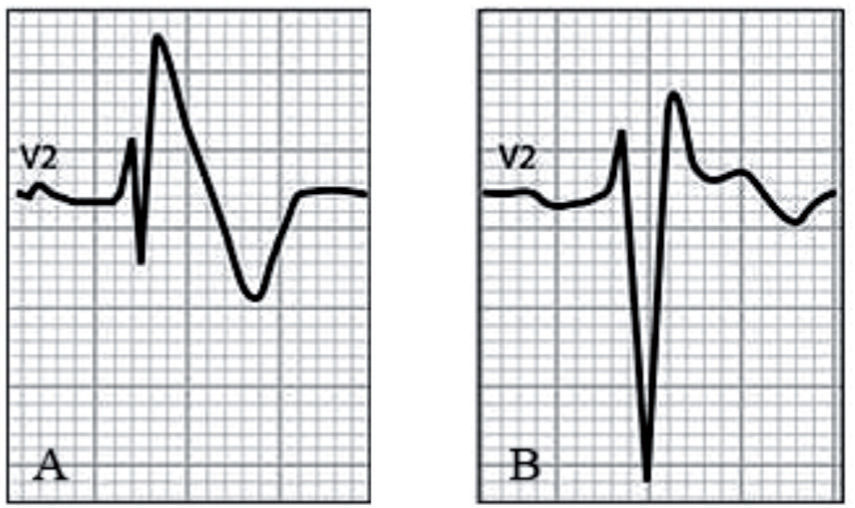
. A) Patrón electrocardiográfico tipo 1. Elevación del ST ≥ 2mm con inversión de la onda T («aleta de tiburón»). B) Patrón electrocardiográfico tipo 2. Elevación del ST ≥ 2mm con onda T isodifásica («silla de montar»).")
Síndrome de Brugada (Corrado D. Recommendations for interpretation of 12-lead electrocardiogram in the athlete. Eur Heart J. 2010;31:243–59). A) Patrón electrocardiográfico tipo 1. Elevación del ST ≥ 2mm con inversión de la onda T («aleta de tiburón»). B) Patrón electrocardiográfico tipo 2. Elevación del ST ≥ 2mm con onda T isodifásica («silla de montar»).
El diagnóstico definitivo de SBr requiere la documentación del patrón ECG tipo 1 de forma espontánea o después de un test de provocación farmacológico con bloqueadores del canal del sodio (flecainida).
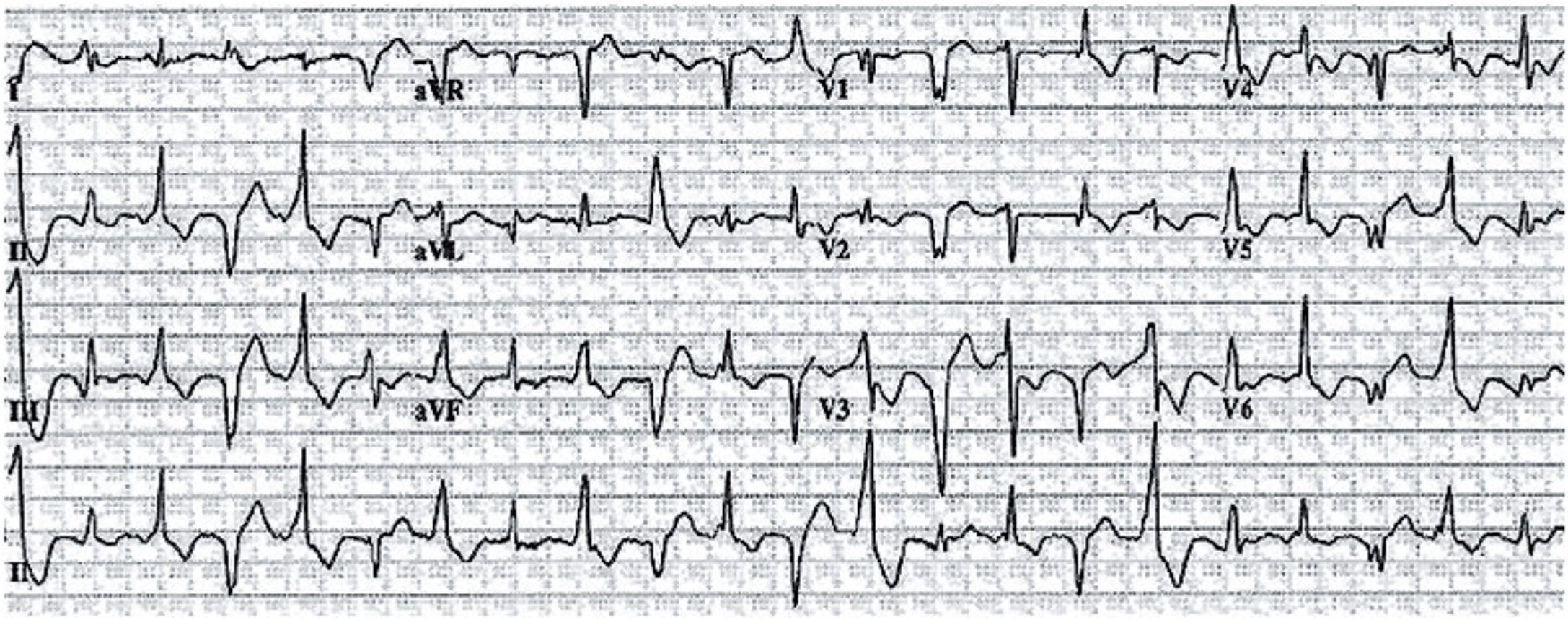
Taquicardia ventricular polimórfica catecolaminérgicaLa taquicardia ventricular polimórfica catecolaminérgica (TVPC) es una canalopatía arritmogénica muy poco frecuente, que se caracteriza por alteraciones en la regulación del calcio intracelular en relación con estímulos adrenérgicos que son sustrato para la aparición de arritmias ventriculares graves y MS en niños y jóvenes31. La edad media de presentación es 7–9 años en forma de síncope y/o MS desencadenados por ejercicio o por emociones intensas. El ECG en reposo es normal pero durante el ejercicio es habitual que se desencadenen extrasístoles ventriculares cuya complejidad aumenta a medida que progresa el ejercicio hasta degenerar en taquicardia ventricular polimórfica y fibrilación ventricular (fig. 16). Aunque existe historia familiar de MS hasta en el 30% de los casos, la elevada mortalidad en etapas tempranas de la vida sugiere que la mayoría de casos son nuevas mutaciones. El diagnóstico se basa en la capacidad para reproducir las arritmias ventriculares durante el esfuerzo.

Taquicardia ventricular polimórfica catecolaminérgica. El registro muestra el electrocardiograma característico en un paciente generalmente menor de 10 años durante una situación emocional intensa. Se observa aparición de una extrasistolia ventricular, en la que el eje de los complejos QRS oscila latido a latido con una rotación de 180°, lo que acaba dando lugar a una taquicardia ventricular «bidireccional», que puede degenerar en taquicardia ventricular polimórfica y fibrilación ventricular
El síndrome de QT corto es una canalopatía hereditaria recientemente descrita, caracterizada por un intervalo QT muy corto y MS en pacientes de cualquier edad32. Es extremadamente infrecuente pero tiene un patrón ECG característico: QTc: < 360ms (rango entre 220 y 360ms); segmento ST muy corto o ausente; ondas T altas, picudas en precordiales, que pueden ser positivas (jóvenes) o negativas (niños) (fig. 17).
 y un QTc de 324ms. Apenas existe intervalo ST y presenta ondas T negativas muy picudas32.")
Detalle del registro electrocardiográfico de paciente de 2 años estudiado por episodio de muerte súbita abortada a los 4 meses y diagnosticado de síndrome de QT corto. Presenta un intervalo QT llamativamente corto (210ms) y un QTc de 324ms. Apenas existe intervalo ST y presenta ondas T negativas muy picudas32.
No cabe duda de que el diagnóstico precoz de las enfermedades potencialmente letales en la infancia conlleva un gran beneficio. Sin embargo, en el momento actual no existen recomendaciones definitivas para la realización de programas de detección precoz de estas enfermedades.
Muchos de los pacientes con riesgo de MS consultan por clínica específica (síncope de esfuerzo) o completamente inespecífica (mareos, palpitaciones, etc.) y en su evaluación el ECG constituye una herramienta habitual. Afortunadamente, la mayor parte de las cardiopatías que causan MS tiene expresión en el ECG basal por lo que el conocimiento de los marcadores ECG puede permitirnos identificar pacientes en riesgo.
Sin embargo, dado que existen cardiopatías que no producen alteraciones en el ECG basal o que, por su carácter dinámico, pueden dar lugar a registros ECG normales en un momento determinado, es necesario seguir «persiguiendo» estas entidades, sobre todo en aquellos casos en que existen antecedentes familiares o en que exista sospecha clínica fundada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.