











Puntos clave

La hipófisis humana es un órgano neuroendocrino complejo constituida por dos porciones que difieren una de la otra en cuanto a origen embrionario, constitución y función denominadas lóbulo posterior y lóbulo anterior.
El lóbulo anterior o adenohipófisis es de origen epitelial. Procede de una evaginación del ectodermo oral. Contiene 5 tipos diferentes de células que fabrican 6 hormonas. Las células somatotropas producen hormona de crecimiento (GH); las tirotropas, tirotropina (TSH); las lactotropas, prolactina (PRL); las gonadotropas, hormonas foliculostimulante (FSH) y luteinizante (LH), y las corticotropas secretan hormona adrenocorticotropa (ACTH). Una vez descargadas hacia la sangre, son llevadas cada una de ellas por el torrente circulatorio a su destino (célula u órgano “diana”), donde ejercerán su función.
El lóbulo posterior o neurohipófisis proviene del suelo del ectodermo neural y está compuesta por la pars nervosa y el infundíbulo superiores. Se almacenan dos hormonas: la vasopresina (ADH) y la oxitocina, sintetizadas previamente por las células nerviosas del hipotálamo.
La patología neuroendocrinológica deriva de la afección de esta zona. Es múltiple, por lo que la redacción de este artículo se centra en el déficit de hormona de crecimiento (DGH), pubertad precoz central (PPC), diabetes insípida central (DIC) y tumores hipotálamo-hipofisarios.
Déficit de hormona del crecimientoEl déficit de GH, aislado o asociado a otras deficiencias hormonales, es una afección de naturaleza congénita o adquirida, que se caracteriza por la ausencia parcial o total de GH, detectable en plasma o suero.
Este síndrome se caracteriza por una combinación de anomalías auxológicas, clínicas, bioquímicas y metabólicas causadas por una secreción de GH baja, con reducción de los factores de crecimiento que dependen de ella1.
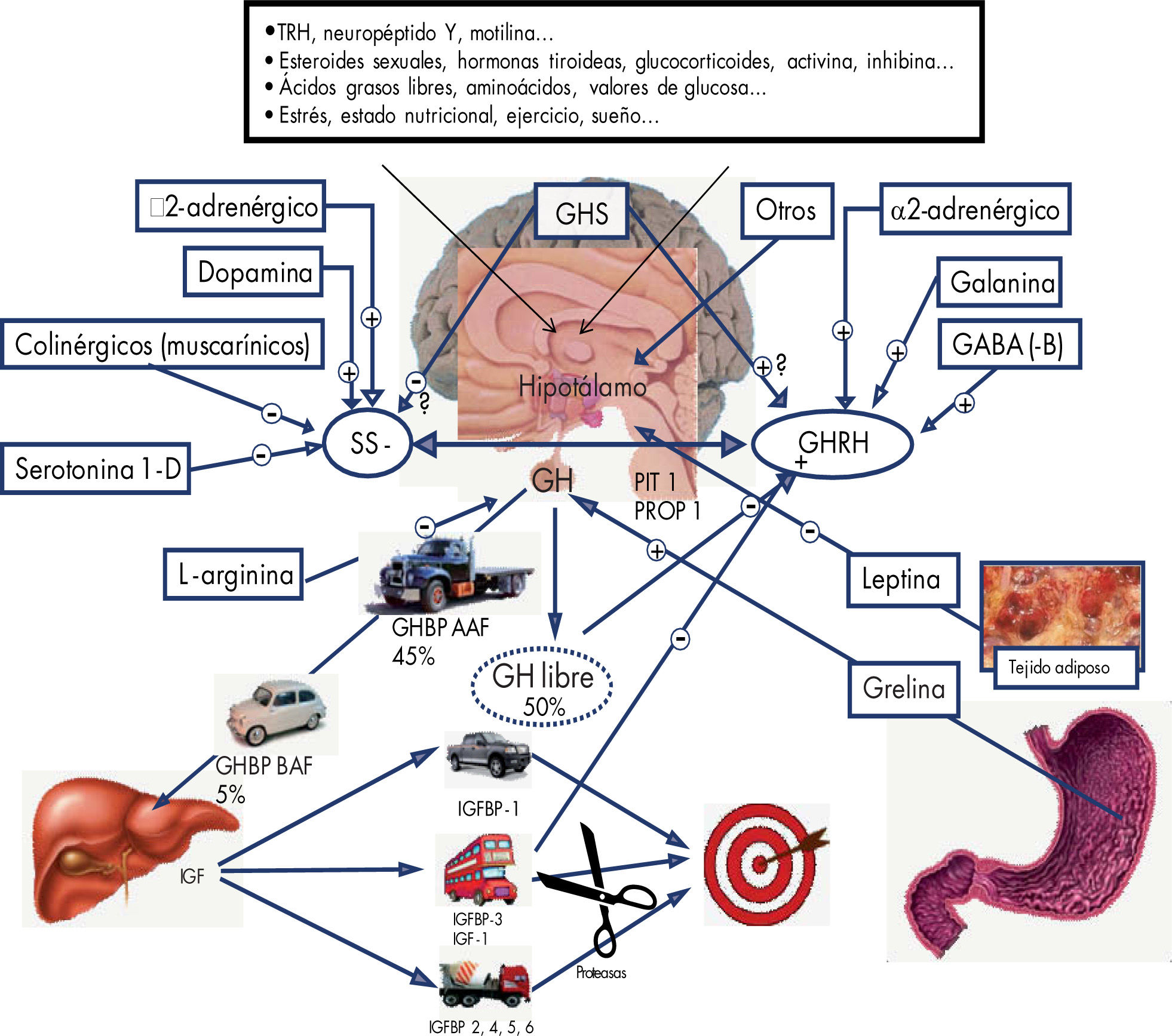
FisiologíaLa producción y liberación de la GH está regulada por dos neurohormonas hipotalámicas: una, denominada factor estimulador de hormona de crecimiento (GHRH), que interviene en la síntesis y liberación de la GH, y otra, la somatostatina (SS o SRIF), que actúa como inhibidor. El control neurogénico central de su secreción es complejo y parcialmente conocido (fig. 1).

Representación esquemática de la secreción y el transporte de la hormona de crecimiento, la liberación de los IFG y el transporte de éstos hasta las células diana. GABA: ácido gamma-aminobutírico; GHBP AAH: proteína transportadora de hormona de crecimiento de alta afinidad; GHBP BAH: proteína transportadora de hormona de crecimiento de baja afinidad; GHRH: hormona liberadora de hormona de crecimiento; GHS: secretagogos de hormona de crecimiento; IGF: factores de crecimiento similares a la insulina; IGFBP:proteína transportadora de factor de crecimiento similar a la insulina; PIT 1 PROT: factores de trascripción específicos de la hipófisis; SS: somatostatina; TRH: hormona liberadora de tirotropina.
Otros factores, como sueño, hipoglucemia, temperatura corporal, ejercicio, ácidos grasos libres o valores de aminoácidos procedentes de la dieta, esteroides sexuales y la leptina, pueden actuar a través de diversos mecanismos sobre la secreción de GH.
La grelina es un péptido de 28 aminoácidos producida mayoritariamente por las células oxínticas del fundus gástrico, aunque también se ha descrito su secreción en el intestino, la hipófisis, el hipotálamo, el riñón, la placenta y el pulmón. Ya se conoce el efecto liberador de GH que tiene la grelina, más potente incluso que el ejercido por la GHRH.
El principal sistema de retrocontrol de la secreción de GH está representado por la somatomedina C o IGF-1.
Se estima que alrededor del 50% de toda de la GH plasmática circulante se encuentra unido a proteínas transportadoras (GHBP), y el resto queda.
La GH ejerce su acción promotora del crecimiento liberando diversos componentes en el hígado, cuyas células hepáticas poseen receptores específicos para aquéllos. A estos mediadores se los conoce como “sistema IGF” (IGF-I e IGF-II, sus receptores y las proteínas transportadoras de los IGF) y las proteasas de las proteínas transportadoras de IGF, que facilitan la liberación del péptido al tejido diana2.
Biología y genética molecular

Los conocimientos sobre biología genética y molecular establecen nuevos horizontes sobre la fisiología de la GH, así como de sus deficiencias. En pocos años, las técnicas moleculares han permitido entender de otro modo los déficits genéticos de la GH, comprender mejor los síndromes de insensibilidad periférica de la GH y diagnosticar nuevas enfermedades endocrinológicas3.
Este trastorno puede ser, a su vez, hipofisario (primario), suprahipofisario (secundario) o bien por resistencia periférica a la GH o a los IFG (periférico).
FrecuenciaEs más frecuente en niños que en niñas, con una relación de 2:1. Generalmente, la incidencia del DGH se estima alrededor de 1/4.000-1/10.000 individuos, aunque probablemente exista una sobrestimación en los datos4.
EtiologíaPuede ser, a su vez, hipofisario (primario), suprahipofisario (secundario) o bien debido a resistencia periférica a la GH (tabla 1).
Causas de deficiencia o de acción de la hormona de crecimiento
| Hipopituitarismo idiopático Síndromes genéticos de la hormona del crecimiento o del GHRH (v. capítulo 20) Síndromes de deficiencia del receptor de hormona de crecimiento (v. capítulo 25) Malformaciones del sistema nervioso central:
|
Infecciones prenatales
Síndromes característicos con anomalías de la línea media Síndrome de ectrodactilia-displasia Ectodérmica-labio leporino
Condiciones con tendencia a malignizaciones frecuentes y que se acompañan de roturas cromosómicas in vitro y defectos inmunitarios
Síndromes que se acompañan de amplias anomalías hipotalámico-cerebrales
|
| Síndromes cromosómicos Otros síndromes Disgenesia hipofisaria Ausencia congénita hipofisaria Hipoplasia hipofisaria Hipófisis ectópica Síndrome de la silla turca vacía Alteraciones de la estructura metabolismo o secreción de la hormona del crecimiento Déficit de GHRH o de su receptor Anomalías del Pit1 y Prop1 Hormona de crecimiento biológicamente inactiva Disfunción neurosecretora de hormona de crecimiento Crecimiento sin hormona de crecimiento Síndrome de hormona de crecimiento invisible Insuficiencia funcional Deficiencia adquirida de hormona de crecimiento
|
Enfermedades inflamatorias del sistema nervioso central
Traumatismos craneales Radioterapia Enfermedades autoinmunitarias
Accidentes vasculares
|
| Trastornos hematológicos |
| Histiocitosis |
| Sarcoidosis |
| Hemocromatosis |
GHRH: hormona liberadora de hormona de crecimiento; Pit: factor de transcripción positivo de la pituitaria; Prot 1: profeta de Pit1.
Lectura rápida
La secreción, transporte y actuación de la hormona de crecimiento (GH) es compleja y parcialmente conocida.
Biología y genética molecularLos avances moleculares han permitido conocer y diagnosticar muchos aspectos de pacientes deficitarios en GH. Asimismo han contribuido al conocimiento de los mecanismos implicados en el eje de la GH.
FrecuenciaSe estima en alrededor de 1/4.000-1/10.000 individuos, aunque probablemente exista una sobrestimación en los datos.
ClínicaEs singular en el periodo neonatal, la lactancia, la infancia y la pubertad, y siempre está presente la pérdida de adquisición de la talla. No se debe de olvidar que en los primeros momentos puede originar hipoglucemias graves.
Depende del momento en que se instaura el déficit en relación con la edad y la fase de crecimiento.
Lectura rápida
Caracterizado por una detención del crecimiento en un paciente que previamente tenía un crecimiento normal.
DiagnósticoNo existe ninguna prueba específica que diagnostique el déficit de GH idiopático, el más común. Se basa en las características clínicas, auxológicas, estudios endocrinológicos y pruebas de imagen. Los estudios moleculares permiten el diagnóstico de deleciones o mutaciones en algunos de los genes implicados.
TratamientoSe hace mediante la administración de GH humana obtenida mediante bioingeniería genética recombinante. El objetivo principal es el incremento de la velocidad de crecimiento y de la talla adulta.
Pubertad precozSe define por la aparición de caracteres sexuales secundarios en niñas menores de 8 años y en niños menores de 9 años.
Tienen un peso y una longitud iguales a los de los recién nacidos normales, debido a la poca relevancia de la GH en el crecimiento longitudinal intrauterino. Es frecuente la ictericia neonatal prolongada. El riesgo obstétrico y perinatal se constata a menudo entre los antecedentes de estos pacientes. En las formas congénitas se pueden producir hipoglucemias en los primeros 4 años. Cuando el déficit de GH se asocia al de ACTH, la hipoglucemia neonatal aparece siempre con cianosis, letargia, convulsiones y shock. Se observa, a veces, microgenitalia (micropene, escroto poco desarrollado, testes pequeños, criptorquidia, hipoplasia de clítoris y labios menores). Pueden tener ausencia de TSH, dando signos y síntomas de hipotiroidismo5.
LactanciaDespués de los 3 meses, y sobre todo cuando han transcurrido 18 meses, empieza a disminuir la velocidad de crecimiento (VC), y a los 2 años se encuentra próximo al percentil 3 de talla o por debajo de él. El desarrollo psicomotor, si no ha habido crisis de hipoglucemia con daño cerebral, es normal.
InfanciaLa VC es lenta, inferior al percentil 25, con lo que la talla se desvía progresivamente de los canales de normalidad; la maduración ósea es inferior en más de un año respecto de la edad cronológica.
Algunos de estos pacientes pueden tener características fenotípicas comunes, lo que les da una apariencia similar: el perímetro cefálico es normal, la facies puede parecer pequeña con frente abombada, raíz nasal aplanada, voz aguda de tono elevado y retraso del brote de la dentición. Se describe como cara de muñeca o de querubín. Hay sobrepeso moderado, acumulación de grasa troncal, aumento de pliegues cutáneos, escaso desarrollo muscular, extremidades de aspecto grácil, con manos y pies pequeños y crecimiento escaso de uñas.
PubertadSi no se ha instaurado tratamiento, la pubertad se suele retrasar, principalmente si existe también un déficit de gonadotropinas.
Déficit de GH adquiridoAquí pueden faltar ciertas manifestaciones de los casos congénitos (p. ej., los rasgos faciales). Al principio, los pacientes muestran un crecimiento normal, enlenteciéndose o deteniéndose después. Esto obliga a descartar una patología orgánica hipotálamohipofisaria.
DiagnósticoEl diagnóstico del DGH en la infancia requiere integrar diferentes aspectos: sintomatología, auxología y valoración fisiológica, pruebas de imagen, genética y morfológica del eje GHIGF1. La evaluación de DGH en el niño de talla baja (talla por debajo de 2 desviaciones estándar [DE] para la media poblacional) no se debería iniciar nunca hasta no descartar todas las causas de fallo de crecimiento. Además, el DGH puede presentarse como un problema aislado o en combinación con deficiencia de otras hormonas hipofisarias.
AuxologíaLos criterios auxológicos que se deben valorar en el DGH son: a) talla baja extrema, más de 3 DE por debajo de la media; b) talla por debajo de 1,5 DE de la talla media parental; c) talla inferior a 2 DE de la media con VC durante un año más de 1 DE por debajo de la media para la edad cronológica, o que exista una disminución de la DE de la talla de más de 0,5 durante un año en niños mayores de 2 años; d) ausencia de talla baja pero velocidad de crecimiento más de 2 DE por debajo de la media durante un año o más de 1,5 DE de forma sostenida durante 2 años; e) signos de lesión intracraneal; f) signos de deficiencia combinada de hormonas hipofisarias, y g) signos y síntomas de DGH en el neonato.
Estudios endocrinológicosNo existe una prueba específica y tampoco ninguna por sí sola concluyente.
La respuesta de GH < 10ng/ml en dos pruebas de secreción puede ser la base del diagnóstico de insuficiencia o déficit de GH. Una prueba de estimulación de GH con respuesta > 10ng/ml continúa siendo recomendada para excluir el diagnóstico de DGH.
Se han desarrollado al menos 34 pruebas de estímulo de secreción de GH, aunque las más usadas son la hipoglucemia insulínica, clonidina, entre ejercicio propranolol, arginina, y glucagón más bloqueadores beta. Todas las pruebas deben realizarse en ayunas6.
Las determinaciones de IGF-1 y IGFBP-3 deberían estar en primera línea de recogida de datos endocrinológicos.
Estudios radiológicosSe deben iniciar con una radiografía de mano y muñeca izquierda, que suele estar retrasada un año o más respecto de la edad cronológica.
La tomografía computarizada selectiva de la silla turca puede informar sobre la hipófisis o calcificaciones, entre otras.
La resonancia magnética del área hipotálamo-hipofisaria es de elección; informa de: neurohipófisis ectópica, ausencia de tallo hipofisario, hipoplasia hipofisaria, agenesia de hipófisis, tumor intracraneal, displasia septoóptica, posibles alteraciones embriológicas y genéticas. Esta técnica contribuye al diagnóstico topográfico.
Estudios molecularesEn la actualidad, puede procederse al diagnóstico de deleciones o mutaciones en los genes de la GH hipofisaria, del factor de transcripción hipofisario numero 1 (Pit 1), el “profeta” del Pit 1 (PROP1), POU1F1, el factor de transcripción HESX 1, el receptor de GH (GHr), el receptor de GHRH (GHRHr) y los IGF. Los recién nacidos con escasa longitud al nacer (< 48cm en niños y < 47cm en niñas), si son obesos, tienen poco pelo y presentan hipoglucemia, deben estudiarse con técnicas moleculares, por si tienen un defecto de la GH o del gen del receptor.
TratamientoLa GH humana obtenida mediante bioingeniería genética recombinante (hrGH) de ADN es el tratamiento de elección. El objetivo es el incremento de la VC y de la talla adulta.
Los datos de estudios clínicos de Europa indican que comenzar con una dosis diaria de 0,025-0,035mg/kg/día se alcanza buen crecimiento en la mayoría de los casos. El tratamiento debe iniciarse de forma temprana cuando esté establecido un diagnóstico de certeza y su retraso puede tener como consecuencia una mala recuperación del crecimiento.
Pubertad precozLa pubertad precoz (PP) se acostumbra a definir por la aparición de caracteres sexuales secundarios en niñas menores de 8 años y en niños menores de 9 años, y por pubertad adelantada cuando aquéllos se manifiestan en edad algo anterior a los límites normales. Así, se establece entre los 10-12 años en los varones y 9-11 años en las mujeres7. Esta no es patológica y suele ser idiopática; si la evolución es lentamente progresiva, no suele afectar a la talla final, pero en otras ocasiones su evolución presenta progresión rápida.
IncidenciaSe calcula en 1/5.000 a 1/10.000 recién nacidos vivos. A menudo es familiar, es más frecuente en niñas respecto a los niños, con relación de 10:1, aunque algunos establecen 23:1. Asimismo, en las niñas se produce más la PP idiopática, mientras que en los varones suelen observarse formas secundarias a un proceso orgánico8.
Clasificación de la pubertad precozSe clasifica en dependientes de la activación de las gonadotropinas, como consecuencia de la actividad de las neuronas hipotalámicas productoras de la hormona liberadora de gonadotropinas (GnRH), lo que conlleva la activación del eje hipotálamo-hipófisisgonadal. Se la denomina también pubertad precoz central (PPC); la segunda forma serían las independientes de las gonadotropinas o PP periférica o seudopubertad precoz (PPP). Se puede establecer una tercera forma de PP denominada parcial, incompleta o variantes del desarrollo normal.
La PPC es siempre isosexual y la PPP puede ser isosexual o heterosexual.
En la tabla 2 se exponen las causas de PP, pero aquí sólo se revisará la PPC.
Causas de pubertad precoz
| Pubertad precoz central, isosexual completa (precocidad sexual dependiente de Gn o activación prematura del generador de pulsos hipotalámico) Idiopática, verdadera
Tumores del SNC
Trastornos del SNC
Exposición crónica previa a esteroides exógenos, hiperplasia suprarrenal congénita, síndrome de McCune-Albright, tumores gonadales o suprarrenales |
| Pubertad precoz periférica, seudopubertad precoz (iso o heterosexual) Síndrome de McCune-Albright (displasia fibrosa poliostótica) Varones
Mujeres
Hipotiroidismo Iatrogénica o exógena (exposición no intencionada a estrógenos de alimentos, fármacos o productos cosméticos) |
| Pubertad precoz parcial Telarquia prematura Pubarquia precoz
|
HCG: gonadotropina coriónica humana; SNC: sistema nervioso central.
Tomado de Cañete e Ibáñez11.
La activación prematura de la secreción pulsátil del factor liberador hipotalámico de gonadotropinas (LH-RH), estimula la secreción de LH y FSH por la hipófisis. Las gonadotropinas estimulan el desarrollo de las gónadas y tiene lugar una pubertad isosexual, con una secuencia de aparición de caracteres sexuales secundarios similar a la pubertad fisiológica, pero con períodos evolutivos más cortos7,9,10.
El cuadro clínico de la PPC se inicia con el desarrollo del botón mamario en niñas y por aumento del volumen testicular en varones (4ml, orquidómetro de Prader). La aparición de vello púbico ocurre varios meses después, aunque a veces pueden coexistir. En ambos sexos, se acompaña de aceleración de la VC11.
Exámenes complementarios en la pubertad precoz centralRadiografía de mano y muñeca izquierdaPara valoración de la maduración ósea que suele estar adelantada respecto de la edad cronológica.
EcografíaEn las niñas, informa del tamaño uterino, la relación cuello/cuerpo, el engrosamiento de endometrio, la forma y el tamaño de los ovarios y la presencia de quistes foliculares o de masas tumorales. Es muy válida, también, para el control del tratamiento. En varones, puede detectar tumores testiculares.
Lectura rápida
Es más frecuente en niñas respecto a los niños.
Clasificación de la pubertad precozSe clasifica en: pubertad precoz central, que siempre es isosexual; pubertad precoz periférica, que puede ser iso o heterosexual, y pubertad precoz parcial.
Pubertad precoz centralSe produce por activación prematura de la secreción pulsátil del factor liberador hipotalámico de gonadotropinas, con liberación de éstas, que estimulan el desarrollo de las gónadas.
Exámenes complementarios en la pubertad precoz centralSe hacen radiografía de mano y muñeca izquierda, ecografía pélvica en las niñas y testicular en los varones, resonancia magnética de hipotálamohipófisis y exámenes de laboratorio en los que destaca la prueba de estimulación de GnRH.
Lectura rápida
Están indicados los análogos superagonistas de factor estimulador de la hormona de crecimiento (GnRH) de liberación lenta.
Diabetes insípida centralEstá causada por un déficit total o parcial de hormona antidiurética (ADH), que se libera en el lóbulo posterior de la hipófisis.
EtiologíaDestacan los tumores hipotálamohipofisarios, histiocitosis, los traumatismos craneoencefálicos y las formas hereditarias autosómicas dominante o recesiva.
ClínicaSe produce polidipsia y poliuria importante.
DiagnósticoSe realiza con la prueba de restricción hídrica bajo supervisión médica y determinación simultánea de ADH. Se completa con pruebas de imagen del cráneo y del hipotálamohipófisis.
Tienen indicación siempre en la PPC del varón y deben ser también de elección en la niña. Permiten conocer algunas patologías silentes durante años, como el hamartoma hipotalámico, la forma y el tamaño de la hipófisis, las malformaciones del sistema nervioso central, los tumores y otras patologías.
Exámenes de laboratorioNo existen criterios unánimes para realizar el diagnóstico de la PPC, que se basa en una síntesis de hallazgos clínicos y determinaciones hormonales dependientes de las técnicas utilizadas en cada laboratorio.
La prueba más acreditada es la estimulación de la FSH y LH con un bolus por vía intravenosa de GnRH a 100mg/m2 con determinaciones a los 20 y 60min. Se ha propuesto como prueba simplificada la administración de GnRH (100mg por vía subcutánea), con una valoración única de LH y FSH a los 40 min12.
Se debe de medir estradiol en las niñas, aunque es poco específico y testosterona en los varones; en estos últimos tienen mayor interés los valores matinales12.
TratamientoTiene un doble objetivo: detener la maduración gonadal y el avance de los caracteres sexuales secundarios, proporcionando al paciente y a su familia mayor bienestar psicológico, y disminuir el avance de la maduración ósea para mejorar la talla adulta.
El análogo de GnRH (triptorelina por vía intramuscular 70-80mg/kg/28 días) es el fármaco de elección y se obtiene una supresión clínica y analítica de la pubertad. Una forma reciente de terapia implica una implantación subcutánea del análogo de hormona de gonadotropina, que da una supresión excelente de liberación de hormona durante 12 meses, pero no se dispone en España13,14.
Lectura rápida
Está indicada la desmopresina y hay varias formas de administración.
Tumores hipotálamohipofisariosEl craneofaringioma tiene gran relevancia en el grupo de tumores hipotálamo-hipofisarios, con repercusión endocrinológica.
ClínicaDepende de la edad, la localización y el tamaño del tumor. Suele haber manifestaciones endocrinológicas, como retraso de crecimiento (talla baja), pubertad retrasada, déficit de hormona de crecimiento, diabetes insípida central, obesidad e hipopituitarismo. Algunos son asintomáticos.
DiagnósticoSe hace por la clínica, la radiología de cráneo y la resonancia magnética hipotálamo-hipofisaria.
TratamientoSe realiza la exéresis quirúrgica seguida de radioterapia. Hay que hacer tratamiento hormonal sustitutivo.

La diabetes insípida central (DIC) o neurógena se caracteriza por la incapacidad para concentrar la orina, con poliuria y polidipsia, y así mantener el equilibrio hídrico. Está causada por un déficit total o parcial de ADH.
EtiologíaTumoresLos craneofaringiomas suelen presentar DIC en el postoperatorio; es infrecuente hallarla antes de la cirugía.
En pacientes con otros tumores del sistema nervioso central en la región hipotalámica o pineal, suelen iniciarse con DIC o PP15, y hasta el 60% de ellos presentan afectación de la hipófisis anterior.
Existen marcadores tumorales de algunos, como la subunidad β de la gonadotropina coriónica humana, alfafetoproteína y fosfatasa alcalina placentaria16,17. El engrosamiento del tallo hipofisario en la resonancia magnética sugiere un germinoma18.
HistiocitosisLa enfermedad de Hand-Schüller-Christian puede presentar DIC, exoftalmos, lesiones osteolíticas, dermatitis seborreica y otitis medias supuradas. En niños, es frecuente la asociación con déficit de GH19.
AutoinmunidadEn las formas idiopáticas, se han encontrado anticuerpos anti-ADH.
Se ha propuesto esta posibilidad por la existencia de anticuerpos anticélulas productoras de ADH20. Se han encontrado en el 75% de los casos de DIC idiopática, pero también en histiocitosis y germinomas, lo que hace dudar de su especificidad.
HerenciaHay formas autosómicas dominante o recesiva21.
Traumatismos y cirugía de tumores selares o supraselaresLos traumatismos craneoencefálicos moderados-severos pueden presentar en la fase aguda DIC transitoria22. La sección alta del tallo hipofisario y el infundíbulo produce DIC transitoria o permanente.
Síndrome de Wolfram o DIDMOAD (diabetes insípida, diabetes mellitus, atrofia óptica e hipoacusia)Es autosómico recesivo producida por la mutación del gen WFS, que codifica una proteína llamada wolframina implicada en varias funciones celulares. En el niño presentan diabetes mellitus, en etapa peripuberal atrofia óptica y DIC después de la segunda década de la vida23.
ClínicaSuele iniciarse de forma brusca, con poliuria y polidipsia, aumento del volumen urinario, incluso nicturia y enuresis en niños que controlaban esfínteres.
DiagnósticoHay que descartar otras causas, como diabetes mellitus, insuficiencia renal, DIC renal, hipopotasemia e hipercalcemia.
Se debe comprobar la poliuria con balance de ingresos y salidas durante 24h, con determinación de la densidad urinaria. Así, se puede descartar la polidipsia primaria o potomanía. Está indicada la prueba de restricción hídrica bajo supervisión médica, con lo que se demuestra la incapacidad para conservar agua.
En la DIC, los valores de ADH son anormalmente bajos, sin incremento en respuesta a la deshidratación.
Al finalizar la prueba de restricción hídrica, se administra desmopresina que deberá provocar aumento de la osmolaridad urinaria (> 450mOsm/l) y disminución de la diuresis.
Se debe completar con una radiografía simple de cráneo (lesiones osteolíticas, calcificaciones, destrucción de la apófisis clinoides posteriores) y una resonancia magnética del cráneo (señal hiperintensa de la neurohipófisis tumores). En las formas idiopáticas, la resonancia magnética se debe hacer cada año, durante varios años, por la posibilidad de ser la primera manifestación de un germinoma.
Hay que hacer una evaluación de las hormonas adenohipofisarias.
TratamientoLa desmopresina (DDAVP) es de elección; es un análogo sintético de la ADH, que se administra por vía oral a 300-400mg/m2 cada 8 h24. También se puede hacer por vía intranasal a 10μg/kg, 1 o 2 veces diarias.
En el postoperatorio inmediato, a 0,02-0,04mg/kg en cuidados intensivos, con control exhaustivo para evitar la intoxicación hídrica.
Tumores hipotálamohipofísariosSon poco frecuentes, con una incidencia anual de 0,1-4,1/100.000 en niños25. Suponen < 4% de los tumores intracraneales.
CraneofaringiomaEs el de mayor relevancia, causante del 80-90% en dicha localización26. Representa el 4-15% de los tumores intracraneales en < 15 años.
Su origen es discutido, aunque se sostiene que proviene de la proliferación de restos de la bolsa de Rathke. El tipo histológico más frecuente en niños es el adamantimoma y suele ser quístico27
ClínicaDepende de la edad, la localización y el tamaño del tumor. Son frecuentes signos de hipertensión intracraneal, cefaleas, vértigo, hidrocefalia, convulsiones, disminución de la agudeza visual y alteraciones del campo visual. Suele haber manifestaciones endocrinológicas, como retraso de crecimiento (talla baja), pubertad retrasada, DGH, DIC, obesidad e hipopituitarismo. Muchos son asintomáticos.
DiagnósticoSe hace con radiografía lateral de cráneo (calcificaciones) y resonancia magnética craneal, que permite la localización del tumor.
TratamientoExéresis quirúrgica, intentando preservar las estructuras vecinas, seguida de radioterapia posquirúrgica, que mejora los resultados.
La recurrencia del craneofaringioma es alta; el 50-75% de los casos desarrolla hipopituitarismo y presentan un riesgo de muerte superior a la población general por desarrollar obesidad, dislipemias y síndrome metabólico, que aumentan el riesgo cardiovascular. Se debe añadir tratamiento sustitutivo hormonal28,29.


