



Puntos clave
- •
Tiene un síndrome pluriglandular o poliglandular autoinmune (SPA) quien presenta más de una enfermedad autoinmune. Se producen por mutación de genes que actúan sobre la inmunidad, con producción de autoanticuerpos que dañan diferentes órganos.
- •
Ante una entidad autoinmune deben analizarse periódicamente autoanticuerpos. Su positividad facilita el diagnóstico precoz de otras enfermedades.
- •
El SPA I se define por tener al menos 2 de 3 entidades principales: candidiasis mucocutánea, hipoparatiroidismo e insuficiencia suprarrenal.
- •
En casi todos los SPA I hay anticuerpos antiinterferón omega, que junto al análisis del gen AIRE facilita el diagnóstico precoz de casos con clínica inhabitual.
- •
Los SPA II y III son infrecuentes en pediatría. Comparten aspectos genéticos y asociación de enfermedades, por lo que a veces son difíciles de diferenciar. La insuficiencia suprarrenal no se presenta en el SPA III.
Los síndromes pluriglandulares o poliglandulares autoinmunes (SPA) son un grupo de enfermedades heterogéneas condicionadas por una alteración en genes que actúan sobre la inmunidad, lo que hace que, junto con factores ambientales no bien conocidos, se produzcan anomalías en la tolerancia inmunitaria, con producción paulatina e incontrolada de autoanticuerpos, que van a dañar diferentes órganos de la economía1,2. Se considera que un paciente tiene un SPA cuando a lo largo de su vida presenta al menos 2 enfermedades de origen autoinmune3.
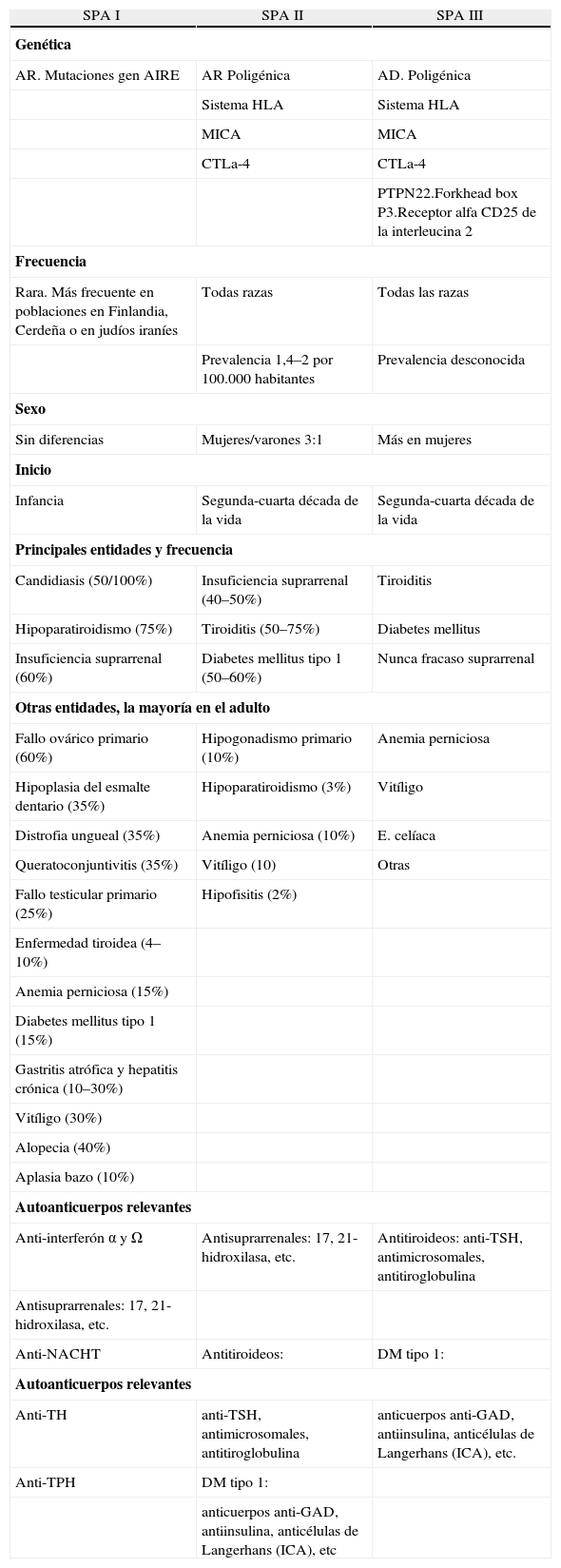
Existen 3 tipos principales de SPA que se llaman I, II y III. Aunque los dos últimos son poco diagnosticados en pediatría, algunas manifestaciones precoces de los mismos pueden aparecer en la infancia, sobre todo en la adolescencia. En la tabla 1 se analizan sus principales diferencias. También se encuadran aquí otras afecciones infrecuentes como los síndromes IPEX, POEMS, Wolfram, o Kearns-Sayre.
Comparación entre los principales síndromes poliglandulares autoinmunes.
| SPA I | SPA II | SPA III |
| Genética | ||
| AR. Mutaciones gen AIRE | AR Poligénica | AD. Poligénica |
| Sistema HLA | Sistema HLA | |
| MICA | MICA | |
| CTLa-4 | CTLa-4 | |
| PTPN22.Forkhead box P3.Receptor alfa CD25 de la interleucina 2 | ||
| Frecuencia | ||
| Rara. Más frecuente en poblaciones en Finlandia, Cerdeña o en judíos iraníes | Todas razas | Todas las razas |
| Prevalencia 1,4–2 por 100.000 habitantes | Prevalencia desconocida | |
| Sexo | ||
| Sin diferencias | Mujeres/varones 3:1 | Más en mujeres |
| Inicio | ||
| Infancia | Segunda-cuarta década de la vida | Segunda-cuarta década de la vida |
| Principales entidades y frecuencia | ||
| Candidiasis (50/100%) | Insuficiencia suprarrenal (40–50%) | Tiroiditis |
| Hipoparatiroidismo (75%) | Tiroiditis (50–75%) | Diabetes mellitus |
| Insuficiencia suprarrenal (60%) | Diabetes mellitus tipo 1 (50–60%) | Nunca fracaso suprarrenal |
| Otras entidades, la mayoría en el adulto | ||
| Fallo ovárico primario (60%) | Hipogonadismo primario (10%) | Anemia perniciosa |
| Hipoplasia del esmalte dentario (35%) | Hipoparatiroidismo (3%) | Vitíligo |
| Distrofia ungueal (35%) | Anemia perniciosa (10%) | E. celíaca |
| Queratoconjuntivitis (35%) | Vitíligo (10) | Otras |
| Fallo testicular primario (25%) | Hipofisitis (2%) | |
| Enfermedad tiroidea (4–10%) | ||
| Anemia perniciosa (15%) | ||
| Diabetes mellitus tipo 1 (15%) | ||
| Gastritis atrófica y hepatitis crónica (10–30%) | ||
| Vitíligo (30%) | ||
| Alopecia (40%) | ||
| Aplasia bazo (10%) | ||
| Autoanticuerpos relevantes | ||
| Anti-interferón α y Ω | Antisuprarrenales: 17, 21-hidroxilasa, etc. | Antitiroideos: anti-TSH, antimicrosomales, antitiroglobulina |
| Antisuprarrenales: 17, 21-hidroxilasa, etc. | ||
| Anti-NACHT | Antitiroideos: | DM tipo 1: |
| Autoanticuerpos relevantes | ||
| Anti-TH | anti-TSH, antimicrosomales, antitiroglobulina | anticuerpos anti-GAD, antiinsulina, anticélulas de Langerhans (ICA), etc. |
| Anti-TPH | DM tipo 1: | |
| anticuerpos anti-GAD, antiinsulina, anticélulas de Langerhans (ICA), etc | ||
AADC: decarboxilasa de los L-aminoácidos aromáticos; NALP5: NACHT, proteína 5 con repetición abundante de leucina; TH: hidroxilasa de la tirosina; TPH: hidroxilasa del triptófano.
Las pruebas de la naturaleza autoinmune de las diferentes enfermedades de los SPA incluyen las siguientes1,4–6:
- –
Muchas de las enfermedades que componen los SPA se asocian a genes que actúan sobre el sistema inmunitario.
- –
Los órganos afectados presentan un infiltrado crónico inflamatorio formado por linfocitos, a veces creando folículos.
- –
Se detectan autoanticuerpos que reaccionan contra antígenos diana específicos de tejidos, enzimas órgano-específicas, productos secretores de las células o receptores celulares.
- –
Dichos autoanticuerpos pueden aparecer tiempo antes de que surjan las manifestaciones clínicas de la enfermedad y predecir su aparición posteriormente.
Cuando un individuo esté afectado de una enfermedad autoinmune determinada, debe realizarse de forma periódica, detección de anticuerpos contra antígenos de glándulas afectadas de un posible SPA. En caso de que fueran positivos, se realizarán estudios de función de esas glándulas, aunque el individuo esté asintomático en ese momento, con el objeto de diagnosticar su fallo precozmente, ya que la presencia de los anticuerpos, como indicadores diagnósticos y marcadores predictivos de una futura enfermedad, está bien establecida. A los pacientes posibles candidatos a presentar un SPA se les debe alertar sobre el hecho de presentar otras enfermedades e instruirles sobre sus síntomas.
Síndrome poliglandular autoinmune tipo I (OMIM 240.300)ConceptoEl SPA tipo I se caracteriza por presentar al menos 2 de las 3 entidades principales que lo componen, la candidiasis mucocutánea crónica, el hipoparatiroidismo y la insuficiencia suprarrenal. Esta tríada clásica aparece en cerca del 60% de los casos 7. Aunque lo habitual es que surjan en el orden descrito, no siempre ocurre así por lo que se han propuesto una serie criterios diagnósticos que se reflejan en la tabla 27.
Criterios para diagnosticar un síndrome poliglandular autoinmune tipo 1.
| Diagnóstico seguro |
| Uno de los siguientes aspectos |
| 1. Presencia de al menos 2 de las siguientes enfermedades: |
| Candidiasis |
| Hipoparatiroidismo |
| Insuficiencia suprarrenal crónica |
| 2. Presencia de una única enfermedad si un familiar tiene un SPA tipo I |
| 3. Una sola enfermedad si se demuestra mutación de ambos alelos del gen AIRE |
| Diagnóstico probables |
| Todos los aspectos siguientes de manera conjunta |
| 1. Presencia de al menos una de las siguientes enfermedades: |
| Candidiasis |
| Hipoparatiroidismo |
| Insuficiencia suprarrenal crónica iniciada antes de los 30 años |
| 2. Presencia de al menos otro componente del síndrome |
| Diarrea crónica |
| Queratitis |
| Exantema periódico con fiebre |
| Estreñimiento severo |
| Hepatitis autoinmune |
| Alopecia |
| Hipoplasia del esmalte dentario |
| 3. Anticuerpos antiinterferón α u Ω |
| 4. Anticuerpos anti-NALP5, AADC, TPH o TH |
AADC: decarboxilasa de los L-aminoácidos aromáticos; NALP5: NACHT, proteína 5 con repetición abundante de leucina; TH: hidroxilasa de la tirosina; TPH: hidroxilasa del triptófano.
Aunque es el SPA menos frecuente, es el más diagnosticado en pediatría, por lo que se le conoce además como SPA tipo juvenil. También se le ha denominado como síndrome de Whitaker, o mediante el acrónimo en inglés APECED, ya que asocia a veces a las entidades descritas una distrofia ectodérmica.
EtiopatogeniaSe hereda casi siempre de manera autosómica recesiva por alteración de un gen de 14 exones llamado regulador autoinmune o AIRE, situado en 21q22.3. Las mutaciones hacen que se codifique una proteína nuclear variante de la normal de 545 aminoácidos que, aunque se expresa en diferentes tejidos, altera sobre todo la función del timo influyendo en la inmunidad celular8–10. Este gen desempeña un papel determinante en la regulación normal del desarrollo de los linfocitos T produciendo autotolerancia inmunitaria. Sus alteraciones permiten una producción incontrolada de anticuerpos contra diferentes tejidos11–15. Se encuentra alterado en el 95% de los casos y se han descrito más 60 mutaciones, aunque las responsables de más del 80% de los casos en pacientes finlandeses y en más del 93% de los de Cerdeña son la R257X7 y la R139X, respectivamente15,16. No obstante, se sospecha que otros genes puedan intervenir en el desarrollo del SPA tipo I.
Lectura rápida
Los síndromes poliglandulares autoinmunes (SPA) son un grupo de enfermedades heterogéneas debidas a una alteración en genes que actúan sobre la inmunidad, lo que hace que, junto con factores ambientales, se produzcan alteraciones en la tolerancia inmunitaria con producción paulatina e incontrolada de autoanticuerpos. Se considera que un paciente tiene un SPA cuando presenta al menos 2 enfermedades autoinmunes.
Es muy rara a nivel global, siendo más habitual en determinadas o poblaciones de Finlandia, Cerdeña o en judíos iraníes. En estos grupos, la frecuencia se ha estimado en un caso cada 9.000 a 25.000 sujetos. Afecta por igual a ambos sexos y aunque lo normal es que se inicie en los primeros años de la infancia, a veces se retrasa su aparición hasta la tercera década de la vida15–22.
Aspectos clínicosLa candidiasis suele verse antes de los 5 años de edad, el hipoparatiroidismo antes de los 10 y la insuficiencia suprarrenal antes de los 15. Otras enfermedades que pueden presentarse en este cuadro son hipogonadismo primario, anemia perniciosa, alopecia y vitíligo, aunque no suelen aparecer en la infancia7,16,22–24.
– Candidiasis mucocutánea. Acontece entre el 50 y el 100% de ellos. En las series finlandesas en cerca del 20% de los casos surge antes del año de edad, en el 50% antes de los 5, en el 70% antes de los 10, en el 90% antes de los 20 y en el 97% antes de los 307.
Afecta sobre todo a la piel, las uñas y las mucosas anal, oral y vaginal, y en menor frecuencia, a la esofágica. La diseminación sistémica es excepcional. Su presencia obliga a realizar el diagnóstico diferencial de inmunodeficiencias congénitas y adquiridas. El diagnóstico se realiza por la clínica y mediante demostración de cándidas en los cultivos realizados.
– Hipoparatiroidismo. El 75% de estos pacientes la presenta. Normalmente, aparece meses o años después de la candidiasis y, generalmente, antes del fracaso suprarrenal. En un tercio de los casos antes de los 5 años de edad y en 2 tercios antes de los 10 años. Es más frecuente en mujeres7.
Se presenta con los signos clásicos de hiperexcitabilidad celular condicionada por la hipocalcemia, como crisis convulsivas, espasmos carpopedales, laringoespasmo y los signos de tetania latente que se objetivan por las pruebas de Chvostek y Trousseau. El electrocardiograma muestra un alargamiento del espacio QT. Desde un punto de vista analítico, encontraremos hipocalcemia, hipomagnesemia, hiperfosforemia y tasas de parathormona (PTH) bajas.
Cuando se asocia a la primera enfermedad comentada, el diagnóstico es fácil de realizar, pero si el hipoparatiroidismo aparece como un hecho aislado, se debe diferenciar del síndrome de Di George, que es debido a una dismorfogénesis de la 3.ª-4.ª bolsas faríngeas y que cursa con hipoplasia del timo y de las paratiroides. También deben descartarse otros cuadros que cursen con hipocalcemia, como los raquitismos y los seudohipoparatiroidismos, pero en ellos la PTH estará elevada.
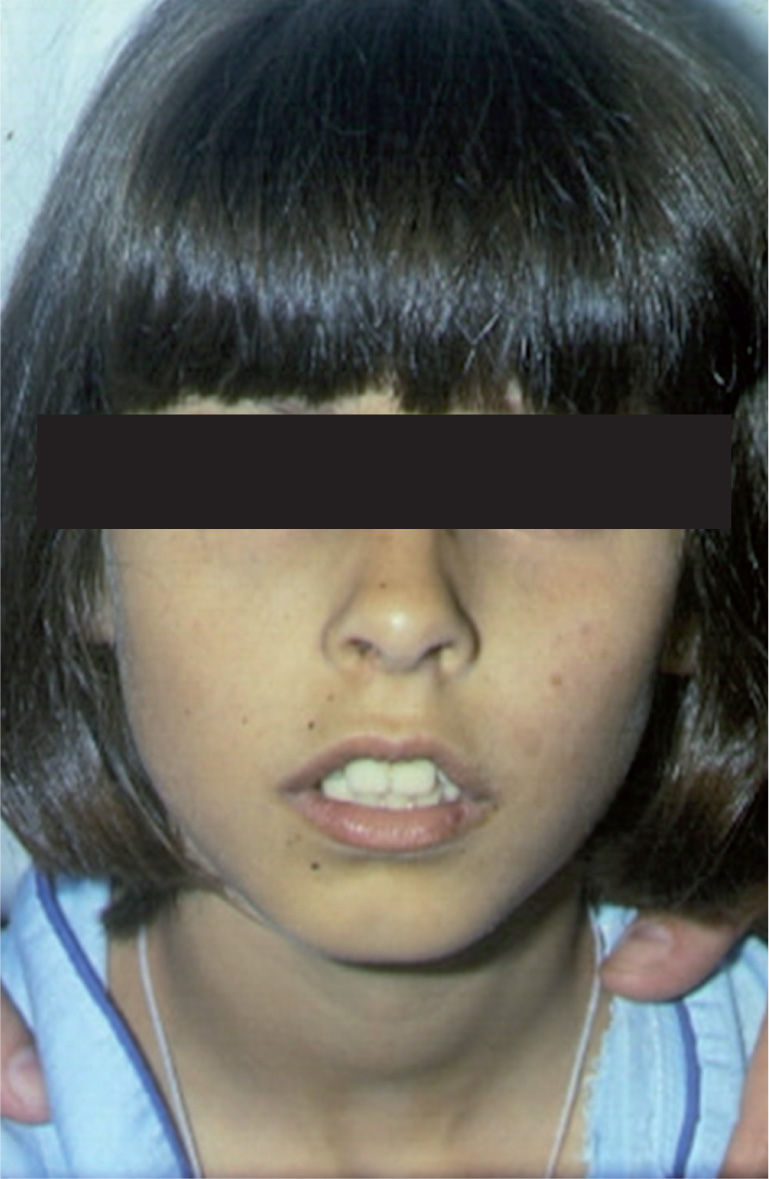
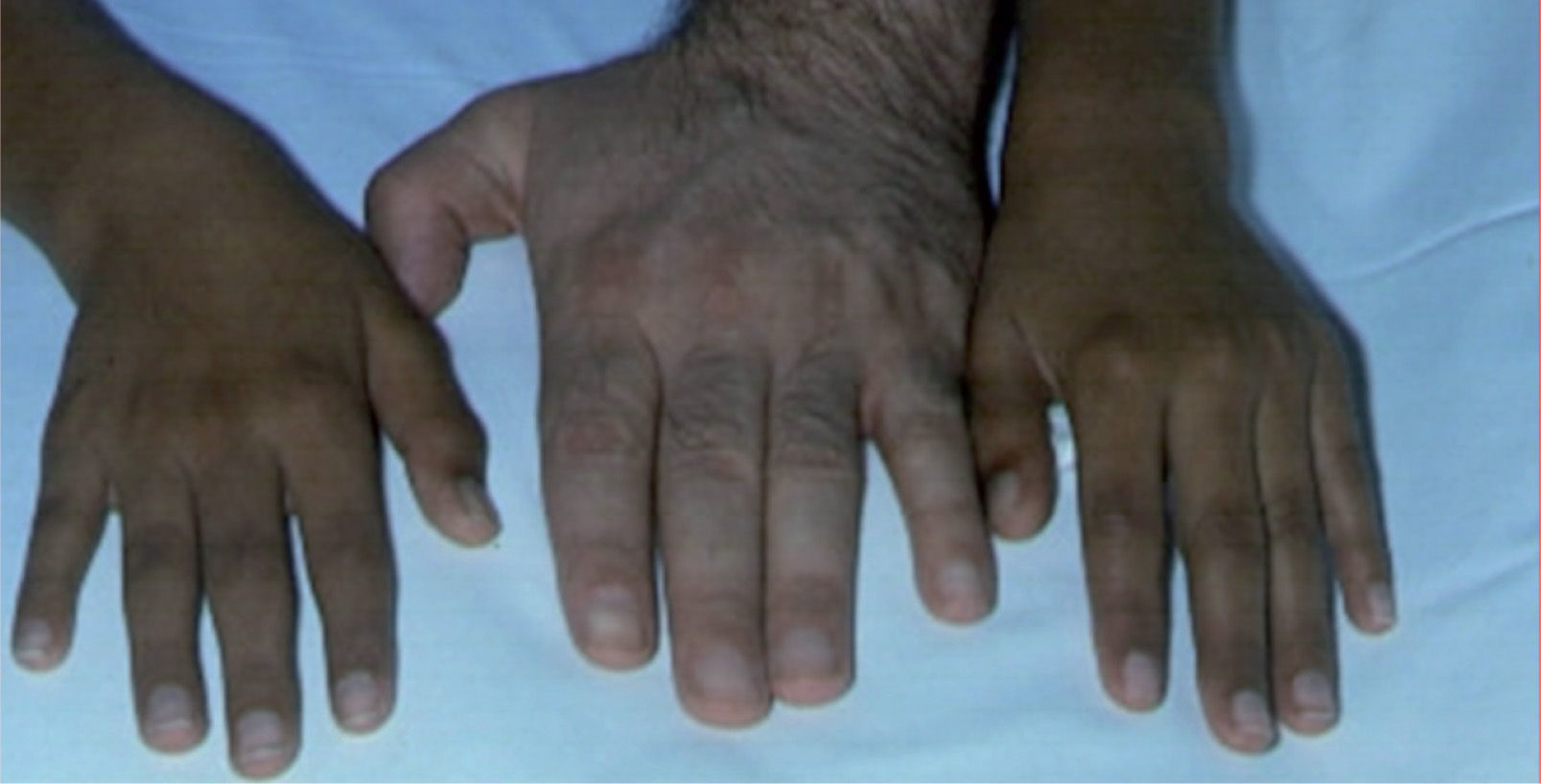
– Fracaso suprarrenal. Ocurre entre los 10 y los 30 años de edad. Se documenta en el 60% de los pacientes. Normalmente, es un fallo total y primario que afecta a la producción de mineral y glucocorticoides, pero a veces se desarrolla de manera parcial, completándose el fracaso global y total en unos 3 años. La expresión clínica se produce gradualmente con signos sutiles, como fatiga, cansancio, hipotensión ortostática, anorexia, vómitos, diarrea e intolerancia al frío, astenia, avidez por la sal, hiperpigmentación cutánea, etc. (figs. 1 y 2). A veces se inicia súbitamente con un cuadro de inestabilidad cardiocirculatoria, deshidratación y un síndrome pierde-sal7. En la analítica hay hiponatremia, hiperpotasemia, hipoglucemia e hipernatriuria. Desde un punto de vista hormonal, existen niveles
Lectura rápida
Cuando un sujeto esté afectado de una afección autoinmunitaria determinada, debe realizarse de forma periódica, detección de autoanticuerpos. Su presencia, como indicadores diagnósticos y marcadores predictivos de una futura enfermedad, está bien establecida, por lo que deben hacerse estudios de función glandular y enseñar a los pacientes la sintomatología de las diferentes enfermedades con el objeto de diagnosticarlas precozmente.
Síndrome poliglandular autoinmume tipo IEl SPA I tiene al menos 2 de las 3 entidades siguientes; candidiasis mucocutánea crónica, hipoparatiroidismo e insuficiencia suprarrenal. La primera suele verse antes de los 5 años de edad, la segunda antes de los 10 y tercera antes de los 15 años. Esta tríada clásica aparece en cerca del 60% de los casos. La candidiasis se detecta entre el 50 y el 100% de los casos, el hipoparatiroidismo en el 75% y el fracaso suprarrenal en el 60%.


– Otras patologías. La hipoplasia del esmalte dentario, la distrofia ungueal y la queratoconjuntivitis aparecen en el 35% de los sujetos. Algunos de ellos presentan de adultos otros cuadros, como hipogonadismo hipergonadotrópico en mujeres en el 60% de los casos, enfermedad tiroidea autoinmune entre el 4 y el 10%, anemia perniciosa en el 15%, diabetes mellitus tipo 1 en el 15%, gastritis atrófica y hepatitis crónica en el 10–20%, asplenia, vitíligo, etc.7. En la serie italiana la hepatitis crónica, estuvo presente en el 27% de los pacientes16.
Protocolos diagnósticosLa presencia de 2 de las 3 afecciones más importantes sigue siendo válida, para diagnosticar un SPA tipo I, pero con ellos se corre el riesgo de no identificar a pacientes en los cuales las manifestaciones clínicas precoces son de entidades menos frecuentes del síndrome, ya que un 11% no tiene como componentes iniciales ninguna de las 3 principales enfermedades7.
La detección de autoanticuerpos contra el interferón Ω y α2, antisuprarrenales o contra NALP5 (NACHT, proteína 5 con repetición abundante de leucina), decarboxilasa de los L-aminoácidos aromáticos (AADC), hidroxilasa del triptófano (TPH) o hidroxilasa de la tirosina (TH) puede facilitar el diagnóstico precoz24,25. En la práctica totalidad de los pacientes se han detectado anticuerpos antiinterferón Ω26–28, mientras los antiparatiroideos como los anti-CASR y anti-PTH, entre el 10 y el 50%. El análisis de anticuerpos anti-NALP5 ha demostrado su utilidad para predecir un futuro hipoparatiroidismo29. También se pueden analizar anticuerpos antisuprarrenales contra la enzima 21-OH-hidroxilasa y 17 hidroxilasa, etc., para diagnosticar precozmente un fracaso suprarrenal30.
En caso de positividad de los anticuerpos o de tener clínica específica de alguna afección glandular concreta, se realizarán pruebas para valorar su función. Para evaluar la de las paratiroides se determinarán periódicamente niveles plasmáticos de calcio, fósforo, PTH y de la corteza suprarrenal, de cortisol, aldosterona, ACTH y renina.
Como lo normal es que lo primero que aparezca es la candidiasis mucocutánea crónica, o cuando existan familiares afectados, se puede realizar el estudio del gen AIRE. Si se encontrara una mutación, el diagnóstico estaría hecho y con mayor razón deberían realizarse las pruebas comentadas anteriormente.
TratamientoLos aspectos clave para tratar estas entidades es anticiparse a su aparición para diagnosticarlas lo más precozmente posible y evitar su morbimortalidad.
– Candidiasis mucocutánea. La mucositis oral debe tratarse rigurosamente para prevenir el futuro desarrollo de neoplasias bucales. Se emplean soluciones por vía oral de nistatina o anfotericina B durante semanas. Otros tratamientos se basan en azoles sistémicos cíclicos administrados de forma oral. Los más utilizados son el fluconazol a las dosis de 3–12mg/kg/día y el itraconazol a 3–10mg/kg/día, una vez al día durante periodos variables de semanas o meses7,31.
– Hipoparatiroidismo. En caso de hipocalcemia grave que produzca una crisis de tetania o convulsiones, se administrará gluconato cálcico por vía intravenosa en 30min a razón de 2mg/kg o 2ml/kg solución al 10% diluida a la mitad en glucosado al 5%,
Lectura rápida
Es muy rara aunque más frecuente en Finlandia, Cerdeña o en judíos iraníes. Se hereda de manera autosómica recesiva por alteración del gen AIRE, situado en 21q22.3, que desempeña un papel determinante en la regulación normal del desarrollo de los linfocitos T, produciendo autotolerancia inmunitaria. Sus alteraciones permiten una producción incontrolada de anticuerpos contra diferentes tejidos del organismo.
La detección de autoanticuerpos contra el interferón Ω y α2, antisuprarrenales, o contra NALP5, AADC, TPH o TH puede facilitar su diagnóstico precoz. En la práctica totalidad de los pacientes se han detectado anticuerpos antiinterferón Ω.
– Fracaso suprarrenal. El tratamiento se hará con glucocorticoides y mineralcorticoides orales. En pediatría, los más empleados son la hidrocortisona a dosis de 9–12mg/m2/día en 2 o 3 tomas diarias, y la 9 alfa-fluorhidrocortisona en dosis única de 0,05 a 0,1mg/día. En situaciones de estrés o enfermedad, las dosis de glucocorticoides se duplican o triplican. Los niños deben llevar una placa de identificación donde diga que presentan una insuficiencia suprarrenal crónica. En caso de fracaso suprarrenal agudo, se hará una reposición de líquidos y electrolitos por vía intravenosa, mediante expansión con suero salino fisiológico (ClNa al 0,9%) a razón de 20ml/kg. Si existe hipoglucemia se añadirá glucosa a dosis de 0,25–0,5g/kg. El tratamiento hormonal se hará con hidrocortisona por vía intravenosa con un bolo inicial de 75–100mg/m2, seguida de bolos de 50–75mg/m2 divididos en 4 dosis33.
Síndrome pluriglandular autoinmune tipo II (OMIM 269.200)ConceptoLa principal característica que define a este cuadro es la presencia de 2 o más glandulopatías autoinmunes, de las cuales las más frecuentes son una adrenalitis asociada a una tiroiditis y/o una diabetes mellitus tipo 1. Otras que pueden aparecer son el hipogonadismo primario, la miastenia gravis y la enfermedad celíaca. También se le denomina síndrome de Schmidt cuando lo que se asocia es la afección suprarrenal y la tiroidea, y de Carpenter cuando lo hacen la adrenalitis, la enfermedad tiroidea y la diabetes mellitus. La aparición de las diferentes afecciones puede diferirse hasta 20 años o más34–36. A diferencia del SPA tipo I, en este cuadro no existe candidiasis mucocutánea crónica.
EtiopatogeniaEs una enfermedad poligénica que se hereda de manera autosómica recesiva con penetrancia incompleta. Se ha asociado con diferentes haplotipos del sistema mayor de histocompatibilidad (HLA), situados en p6, siendo los más frecuentemente implicados los alelos que codifican moléculas de clase II, como el HLA-DR3 y/o DR4 B8. DQ y DR, DRB1*04/DQA1*0301/DQB1*0302 que están implicados en el procesamiento de los antígenos por los linfocitos T y su presentación a determinadas células. Otros genes
Lectura rápida
Es el más frecuente de los SPA pero se diagnostica poco en pediatría, ya que las enfermedades glandulares se suelen desarrollar tras la segunda década de la vida. Las mujeres se afectan en una proporción de 3:1. La principal característica que define a este cuadro es la presencia de 2 o más glandulopatías autoinmunes, de las cuales las más frecuentes son una adrenalitis asociada a una tiroiditis y/o una diabetes mellitus tipo 1.
Es una entidad poligénica que se hereda de manera autosómica recesiva con penetrancia incompleta. Se ha asociado con genes del sistema HLA y otros como el MICA, el CTLa-42 y el PTPN22.
La patogenia del cuadro no es bien conocida. Existen un grado de susceptibilidad genética y factores precipitantes ambientales que pondrían en marcha la producción de autoanticuerpos, lo que induciría la puesta en marcha de fenómenos inflamatorios que destruirían la anatomía y función de las glándulas afectadas. Lo que se hereda es la predisposición a presentar la enfermedad y no la enfermedad en sí.
La patogenia del cuadro no es bien conocida. Se sabe que existe un grado de susceptibilidad genética y que existirían precipitantes ambientales no bien conocidos como virus, proteínas de la leche de vaca, etc., que pondrían en marcha la producción de autoanticuerpos y que mediante fenómenos inflamatorios destruirían la anatomía y la función de las glándulas afectadas. Lo que se hereda es la predisposición a presentar la enfermedad y no la enfermedad en sí.
FrecuenciaEs el más frecuente de todos los SPA; sin embargo, se diagnostica poco en pediatría, ya que las enfermedades glandulares se suelen desarrollar a partir de la tercera década de la vida. No obstante, las enfermedades más precoces pueden verse en la adolescencia. Las mujeres se afectan en una proporción 3:1 respecto a los varones. Afecta a todas las razas por igual y su prevalencia se ha estimado en 1,4–2 por 100.000 habitantes34–36.
Aspectos clínicosEl aspecto clave para diagnosticar este proceso es la presencia de una adrenalitis autoinmune que se presente en cualquier época de la vida, ya que suele ser la primera glandulopatía en aparecer. Se ha cifrado que el 50% de los pacientes con insuficiencia suprarrenal de origen autoinmune desarrollarán otra enfermedad de esa naturaleza en el futuro. Por otra parte, entre el 40 y el 50% de los pacientes con SPA tipo II presentan en algún momento insuficiencia suprarrenal34,35.
Alrededor del 50 al 60% de los pacientes con SPA tipo II presentan diabetes mellitus tipo 1, a veces también como primera manifestación del cuadro. Se estima que hasta el 25% de ellos pueden desarrollar posteriormente otras enfermedades autoinmunes36.
La afectación tiroidea autoinmune como hecho aislado es muy frecuente en la población general, pero las posibilidades de asociación entre una tiroiditis que aparece primero y una adrenalitis se han estimado en solo el 1% de esos pacientes. La tiroiditis autoinmune se da en el 50 a 75% de estos pacientes. Las principales asociaciones son diabetes mellitus y tiroiditis en el 40% de los pacientes, tiroiditis y adrenalitis en el 15%, y tiroiditis, diabetes y adrenalitis en el 3%. Otras asociaciones, como diabetes mellitus y vitíligo, o tiroiditis y vitíligo, se han cifrado en el 10%6,36.
– El fracaso suprarrenal. La expresión clínica se comenta en el texto del SPA tipo I.
– La diabetes mellitus tipo 1 se presenta con los clásicos síntomas cardinales, como son poliuria, polidipsia polifagia, pérdida de peso, etc.; encontraremos cifras de glucosa en sangre elevadas y glucosuria; también puede presentarse como una cetoacidosis diabética, con un cuadro clínico de deshidratación, respiración de Kussmaul, etc.; en la analítica encontraremos hiperglucemia y cetosis, con una acidosis metabólica con hiato aniónico elevado.
– La tiroiditis. De manera aislada o en el seno de un SPA, puede cursar con normofunción tiroidea, con hipotiroidismo o con hipertiroidismo. El hallazgo clínico que se encuentra con más frecuencia es un bocio, que se suele detectar en una exploración física rutinaria37.
La hipofunción tiroidea se presenta con síntomas sutiles como intolerancia al frío, retraso de crecimiento, estreñimiento, piel seca y alteración del rendimiento escolar. En caso de hipertiroidismo, encontraremos síntomas cardiocirculatorios, como taquicardia e hipercinesia circulatoria; alteraciones del sistema nervioso central y periférico, como trastornos vasomotores, labilidad emocional, irritabilidad, llanto inmotivado, distracción, mal rendimiento escolar, y trastornos del sueño, temblor e intolerancia al calor, etc. Otros signos y síntomas son exoftalmos y diarrea, pérdida de peso, etc. Las tormentas tiroideas son excepcionales en pediatría.
Protocolos diagnósticosSe realizarán determinaciones periódicas de autoanticuerpos; los antisuprarrenales ya han sido comentados; los antitiroideos son los antihormona tiroestimulante (TSH), antimicrosomales, antitiroglobulina, etc., y para la diabetes mellitus tipo 1, los antidecarboxilasa del ácido glutámico (GAD), antiinsulina, anticélulas de Langerhans (ICA), etc.1. En caso de positividad de ellos o de tener clínica específica de alguna afección glandular, se realizarán pruebas para valorar su función. Para analizar la función tiroidea se determinarán los niveles de TSH, T4-L, etc. En caso de sospechar diabetes mellitus,
Lectura rápida
Los límites entre el SPA tipo II y III no están bien definidos desde un punto de vista clínico y en relación con su etiopatogenia. Lo que les diferencia es la ausencia de una insuficiencia suprarrenal en el SPA III. Asocia también 2 enfermedades de origen autoinmunitario. Lo más frecuente es presentar una enfermedad tiroidea y alguna otra. Si lo hace junto a una diabetes mellitus se denomina tipo III A. Si es junto a una anemia perniciosa o a una gastritis atrófica, se denomina III B, y si lo hace con vitíligo, alopecia o miastenia gravis, se llama III C.
Su frecuencia es desconocida, es similar en ambos sexos, afecta a todas las razas y, normalmente, a adultos. Está condicionado genéticamente e influido por factores ambientales. Se hereda de manera autosómica dominante con penetrancia incompleta. Se ha asociado a los mismos genes del SPA II y a otros exclusivos como el denominado forkhead box P3 en el cromosoma X y el receptor alfa CD25 de la interleucina-2 en el 10.
Las situaciones que cursen con normofunción tiroidea solo precisarán monitorización periódica. Se tratarán los casos con hipofunción tiroidea o con hipertiroidismo. En el primer caso se administrará levotiroxina oral a dosis 1–3μmg/kg/día para conseguir restaurar la función tiroidea. En el segundo caso existen 3 posibilidades terapéuticas: los fármacos antitiroideos por vía oral, el radioyodo y la cirugía. De las primeras, las más utilizadas son el propiltiouracilo a razón de, 5–10mg/kg/día y el metimazol o el carbimazol a 0,5–1mg/kg/día37,38.
El tratamiento de la diabetes mellitus consiste en pautas de autocontrol, ejercicio físico, actuaciones dietéticas y administración de insulina subcutánea. El tratamiento del fracaso suprarrenal se comenta en el texto del SPA tipo I.
Síndrome poliglandular autoinmune tipo IIILos límites entre el SPA tipo II y III no están bien delimitados desde un punto de vista clínico y en relación con la etiopatogenia. Lo que les diferencia es la ausencia de una insuficiencia suprarrenal en el SPA tipo III. Asocia también 2 enfermedades de origen autoinmunitario. Lo más frecuente es presentar una enfermedad tiroidea autoinmune y alguna otra. Si lo hace junto a una diabetes mellitus, se denomina tipo III A; si es junto a una anemia perniciosa o a una gastritis atrófica, se denomina III B, y si lo hace con vitíligo, alopecia o miastenia gravis, se llama III C. La frecuencia de este síndrome es desconocida. Afecta a todas las razas, es similar en ambos sexos y normalmente afecta a adultos, aunque se han descrito casos infantiles; los más jóvenes han sido un niño de 12 años con vitíligo, alopecia y tiroiditis autoinmune, y otro de 8 años que asociaba deficiencia de hormona de crecimiento (GH), hipertiroidismo y diabetes mellitus39,40.
La naturaleza autoinmunitaria de esta enfermedad está condicionada genéticamente e influida por factores ambientales, como infecciones virales, introducción precoz de la leche de vaca o administración de interferón-α para tratar la hepatitis C. Parece heredarse de manera autosómica dominante con penetrancia incompleta. Se ha asociado con los mismos genes relatados en el caso del SPA tipo II y con otros exclusivos, como el denominado forkhead box P3 en el cromosoma X, y el receptor alfa CD25 de la interleucina 2 en el cromosoma 1041. También se ha detectado la presencia de autoanticuerpos relacionados con la tiroiditis, la diabetes mellitus y otras enfermedades autoinmunitarias.
- •
Las principales enfermedades de este síndrome son la tiroiditis en todas sus vertientes de normofunción, hipofunción o hiperfunción.
- •
La diabetes mellitus tipo, 1 como se ha descrito.
- •
Anemia perniciosa, vitíligo y alopecia rara vez se ven en la infancia.
- •
El tratamiento de las entidades más importantes de este cuadro se ha relatado anteriormente.
Conocido como síndrome de inmunodisregulación, con enteropatía y poliendocrinopatía ligado al cromosoma X. Es muy raro, cursa con diarrea incoercible, dermatitis ictiosiforme, diabetes mellitus tipo 1, trombocitopenia, anemia hemolítica, enfermedad tiroidea neonatal, hepatitis y nefritis. Afecta a lactantes, su curso suele ser fatal y el único tratamiento son los inmunosupresores o el trasplante de médula ósea. Se debe a una proliferación sin control de diferentes linfocinas y es producido por una mutación del gen FOXP3, situado en Xp23, que condiciona alteraciones en la proteína escurfina, que interviene en el desarrollo de células T reguladoras. Se ha encontrado atrofia de vellosidades, infiltrados linfocitarios en la lámina propia, anticuerpos antienterocitos y contra otros tejidos. En algunos pacientes se halló en la necropsia ausencia de islotes de Langerhans42.
Síndrome de POEMS o de Crow-Fukase (OMIM 192.240)Es un trastorno infrecuente multisistémico que cursa con polineuropatía y endocrino
Lectura rápida
Son muy infrecuentes, como el IPEX, que cursa con enteropatía y poliendocrinopatía ligado al cromosoma X; el POEMS, que tiene una polineuropatía y endocrinopatías diversas; el DIDMOAD con atrofia óptica y diabetes mellitus tipo 1, diabetes insípida y sordera, y el KEARNS-SAYRE, que asocia oftalmoplejía, retinitis pigmentaria y cardiomiopatía.
Cursa con atrofia óptica y diabetes mellitus tipo 1 como criterios mayores, y a veces diabetes insípida y sordera (DIDMOAD). Es causado por una mutación de un gen situado en 4p, que codifica una proteína denominada wolframina. Se hereda de manera autosómica recesiva, aunque una variante se considera una enfermedad mitocondrial44.
Síndrome de Kearns-Sayre (OMIM 530.000)Asocia oftalmoplejía, retinitis pigmentaria y cardiomiopatía. Se considera una enfermedad mitocondrial por alteración de genes de la cadena respiratoria. En biopsias musculares se ven fibras rojo-rasgadas. Las principales endocrinopatías son la deficiencia de GH, la diabetes mellitus y el hipoparatiroidismo45.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.