














Puntos clave

Las enfermedades del túbulo renal se caracterizan por alteraciones específicas de la función tubular, sin disminución primaria del filtrado glomerular (FG).
El ultrafiltrado glomerular se modifica a su paso por los túbulos renales mediante complejos mecanismos de reabsorción y secreción para su transformación en orina. La intervención tubular es fundamental para adaptar la excreción de agua y solutos en función de la dieta y la producción endógena.
En la nefrona proximal, se produce la reabsorción del mayor porcentaje de agua y solutos filtrados, mientras que en la nefrona distal tiene lugar el ajuste final de la excreción de estas sustancias, de la osmolaridad urinaria y la regulación del equilibrio ácido-base1–3.
Las tubulopatías se pueden clasificar en:
- —
Simples y complejas, según se altere uno o varios mecanismos de transporte tubular.
- —
Primarias-congénitas y secundarias (a tóxicos o a otras enfermedades sistémicas, urológicas o renales adquiridas).
Las tubulopatías son enfermedades poco frecuentes y pueden manifestarse de forma heterogénea. Se requiere una interpretación correcta de la anamnesis, así como de los hallazgos obtenidos de la exploración física y la analítica de sangre y orina para llegar a plantearse la sospecha diagnóstica.
Frecuentemente, se trata de pacientes con manifestaciones clínicas inespecíficas, con retraso en el desarrollo ponderoestatural, anorexia, vómitos, estreñimiento, irritabilidad y poliuria con deshidrataciones frecuentes; además, es habitual la alteración hidroelectrolítica y del equilibrio ácido-base. A continuación se enumeran las posibles formas de presentación4:
- —
Antecedentes perinatales de riesgo (prematuridad, bajo peso al nacer, polihidramnios).
- —
Afectación general con astenia, malestar e irritabilidad, hipertermia.
- —
Avidez por la sal y deshidratación.
- —
Poliuria. Polidipsia.
- —
Síntomas digestivos: anorexia, vómitos, estreñimiento, dificultad para la alimentación.
- —
Tetania. Raquitismo.
- —
Anomalías oculares e/o hipoacusia.
- —
Retraso ponderoestatural.
- —
Alteraciones electrolíticas.
- —
Alteraciones en el equilibrio ácido-base.
- —
Litiasis renal y/o nefrocalcinosis.
- —
Infección del tracto urinario.
En la anamnesis, debemos recoger los antecedentes familiares de enfermedad tubular y la posible consanguinidad, ya que algunas son enfermedades con herencia autosómica recesiva (AR), así como los antecedentes obstétricos y neonatales, la historia dietética (avidez por la sal, ingesta de líquidos) y el ritmo de diuresis (valorar nicturia y enuresis). Es importante investigar acerca de la administración de fármacos, tóxicos o de la presencia de otras enfermedades concomitantes.
En la exploración física, debemos realizar una exploración completa por aparatos (signos de raquitismo y alteraciones oculares y/o auditivas que pueden asociarse), valorando globalmente los estados de hidratación, nutricional y de crecimiento y desarrollo. Se recogerá el peso, la talla y la presión arterial.
El estudio básico ante la sospecha de afectación tubular debe incluir5,6:
- —
Estudio del equilibrio hidrosalino con la determinación de iones (sodio [Na], potasio [K], cloro [Cl], calcio [Ca], fósforo [P], magnesio [Mg]) en sangre y orina simultáneamente, de las excreciones en orina minutada de 24h y los índices en micción, así como las excreciones fraccionales (EF) o los índices de excreción (IE).
- a)
EFNa (%): 0,6 ± 0,35
- b)
EFK (%): 9,6 ± 4
- c)
EFCl (%): similar a la de sodio, ligeramente más elevado.
Lectura rápida
IntroducciónLas enfermedades del túbulo renal son un conjunto de enfermedades caracterizadas por alteraciones específicas de la función tubular con filtrado glomerular inicial normal.
Pueden ser primarias, de origen genético, o secundarias a otras enfermedades o a fármacos y tóxicos. Actualmente, dados los avances sobre las bases moleculares, podemos conocer el sustrato genético de muchas de estas enfermedades.
Siempre deben descartarse, además, alteraciones de la vía urinaria y nefropatías con componente intersticial.
Enfoque diagnósticoLa sintomatología clínica es inespecífica, con retraso en el desarrollo ponderoestatural, anorexia, vómitos, estreñimiento, irritabilidad y poliuria con deshidrataciones frecuentes; son habituales las alteraciones hidroelectrolíticas y del equilibrio ácido-base.
En la anamnesis, debemos recoger los antecedentes familiares de enfermedad tubular. Es importante determinar la ingesta de líquidos, el ritmo de diuresis, y si hay o no avidez por la sal.
Estos parámetros nos ayudan a valorar el manejo de agua y electrolitos por el riñón, así como determinar las cantidades de lo filtrado que finalmente se excretan.
- a)
- —
Para valorar la reabsorción de fósforo, se utiliza la tasa de reabsorción tubular de fosfato (RTP = 1 – EFP), que en lactantes debe ser 78,7 ± 8,4%, y en niños mayores de 2 años, 92 ± 4,2%.
- —
El gradiente transtubular de potasio (GTTK) aporta información acerca de la acción de la aldosterona: (GTTK: [Ko+/(osmolaridado/osmolaridads)1/Ks). Valores inferiores a 4–5 asociados a hiperpotasemia indican hipoaldosteronismo o seudohipoaldosteronismo. También puede ser necesario determinar la renina y la aldosterona plasmáticas.
- —
Estudio del equilibrio ácido-base: pH en sangre y orina.
- —
La tira reactiva de orina y el análisis del sedimento urinario nos van a permitir conocer el pH (normal < 5,5) y la densidad urinarios, así como la presencia de proteinuria, glucosuria y de cristales característicos.
- —
Ante el hallazgo de proteinuria, debemos diferenciar la proteinuria tubular o de bajo peso molecular (β2-microglobulina, proteína ligadora del retinol, α1-microglobulina y lisozima) de la glomerular, realizando un perfil proteínico en orina de 24h.
- —
Estudio de aminoácidos en orina.
- —
Estudio de la capacidad de concentración con la evaluación del volumen de orina en 24h. Este es uno de los indicadores más importantes de función tubular. La poliuria (> 2l/24h/1,73 m2) con episodios frecuentes de deshidratación y disminución del espacio extracelular con/sin pérdida salina son situaciones frecuentes. Debemos analizar también la osmolaridad en sangre y orina como primera aproximación; pero para descartar un déficit en la capacidad de concentración urinaria, es preciso realizar el test de DDAVP (1-desamino-8-D-arginina-desmopresina).
- —
Para completar el estudio del metabolismo fosfocálcico, se realizará además la determinación de la parathormona (PTH) y de metabolitos de la vitamina D, radiografías óseas para valorar raquitismo y densitometría.
- —
En ocasiones, es preciso realizar pruebas funcionales para afinar el diagnóstico.
- —
Otras pruebas complementarias que pueden ser necesarias son la ecografía renal, la audiometría, el estudio oftalmológico y el estudio genético.


Ante la sospecha de tubulopatía, deberán descartarse alteraciones de la vía urinaria (displasia, hipoplasia, uropatías graves) o nefropatías con disminución del FG (nefronoptisis, insuficiencia renal crónica) en las que también se produce alteración en la capacidad de concentración de la orina.
En ocasiones, procesos como una gastroenteritis aguda pueden descompensar el equilibrio homeostático en estos pacientes y desenmascarar posibles alteraciones de base.
Principales tubulopatías primariasLas distintas entidades se manifestarán con alteraciones características según el segmento de la nefrona donde se localice la disfunción. Las tubulopatías se pueden clasificar según el/los tipo/s de transportador/es alterado/s (tabla 1). Otra clasificación útil desde el punto de vista didáctico es la que diferencia las anomalías en 3 grupos según la localización en la nefrona (tabla 2).
Clasificación de las tubulopatías según el tipo de transporte alterado
| Trastorno del transporte de proteínas |
| Proteinuria tubular |
| Trastorno del transporte de glucosa |
| Glucosuria renal hereditaria |
| Trastorno del transporte de aminoácidos |
| Cistinuria |
| Hipercistinuria aislada |
| Hiperaminoaciduria dibásica (tipo I, tipo II, lisinuria) |
| Enfermedad de Hartnup |
| Hipoabsorción de metionina |
| Histidinuria |
| Iminoglicinuria familiar y otras glicinurias |
| Aminoaciduria dicarboxílica |
| Aminoacidurias renales no específicas |
| Trastorno del transporte de fosfatos |
| Hipofosfatemia familiar |
| Seudohipoparatiroidismo |
| Trastorno del transporte de ácido úrico |
| Hipouricemia de origen renal |
| Trastorno múltiple del túbulo proximal |
| Síndrome de Fanconi |
| Trastornos de la función reguladora del equilibrio ácido base |
| Acidosis tubular renal |
| Trastorno del transporte de calcio |
| Hipercalciuria de origen renal |
| Trastorno del transporte de sodio, potasio y magnesio |
| Seudoaldosteronismo tipo I |
| Seudoaldosteronismo tipo II o síndrome de Gordon |
| Síndrome de Bartter |
| Síndrome de Gitelman |
| Síndrome de Liddle |
| Déficit de 11 β-hidrosteroide deshidrogenasa |
| Hipomagnesemia-hipercalciuria familiar |
| Trastorno del transporte de agua |
| Diabetes insípida nefrogénica hereditaria |
Clasificación de las tubulopatías según la zona de la neurona afectada
| Alteraciones de la nefrona proximal |
| Síndrome de Fanconi |
| Acidosis tubular renal proximal o tipo II |
| Raquitismo hipofosfatémico ligado al cromosoma X |
| Raquitismos dependientes de la vitamina D |
| Glucosuria renal |
| Aminoacidurias |
| Hipouricemia renal congénita |
| Cistinosis |
| Nefrolitiasis hipercalciúrica ligada al cromosoma X |
| Alteraciones en el asa de Henle |
| Síndrome de Bartter |
| Síndrome de Gitelman |
| Hipomagnesemia familiar con hipercalciuria y nefrocalcinosis |
| Alteraciones de la nefrona distal |
| Acidosis tubular renal distal o tipo I |
| Acidosis tubular renal hipercaliémica o tipo IV o seudohipoaldosteronismo |
| Seudohiperaldosteronismo |
| Diabetes insípida nefrogénica hereditaria |
A continuación se describen algunas de las enfermedades más representativas.
Síndrome de Toni-Debré-Fanconi (tubulopatía proximal compleja)Consiste en una disfunción generalizada del túbulo proximal que determina aumento de la excreción urinaria de fosfato, bicarbonato, sodio, potasio, aminoácidos, proteínas de bajo peso molecular y glucosa. Se manifiesta, por tanto, clínicamente como glucosuria con poliuria, acidosis (acidosis tubular renal [ATR] proximal), deshidratación, hipopotasemia, hipofosfatemia, raquitismo resistente a la vitamina D, anorexia y vómitos intermitentes, debilidad muscular y fallo de medro. También se producen cuadros incompletos en los que la aparición de las manifestaciones clínicas puede ser evolutiva. Dado que se trata de una alteración múltiple, se plantea que podría deberse a la disrupción de una fuente importante de energía, a la alteración inespecífica de la permeabilidad celular o a enfermedad en organelas intracelulares.
Puede ser hereditario (primario o en contexto de errores innatos del metabolismo) o adquirido (tóxicos, inmunológico) (tabla 3); transitorio o irreversible según el agente etiológico, por lo que también varía mucho la edad de presentación. Ante alteraciones clínicas compatibles con síndrome de Fanconi, siempre se deben descartar las formas secundarias7.
Etiología del síndrome de Fanconi
| Origen genético |
Idiopático
|
Asociado a enfermedades genéticas
|
| Origen adquirido |
Asociado a enfermedades adquiridas
|
Asociado a medicamentos y tóxicos
|
- —
El específico de la causa, si se conoce.
- —
Aportes de cloruro sódico, potasio, fósforo y bicarbonato/citrato por vía oral.
- —
Metabolitos de la vitamina D.
- —
Libre acceso al agua.
- —
Los aportes intravenosos pueden ser necesarios al diagnóstico y en descompensaciones por enfermedades intercurrentes. Es importante realizar rehidratación de forma temprana en las situaciones en que se incremente la pérdida de líquidos y electrolitos, como pueden ser los episodios de gastroenteritis aguda o en procesos febriles.
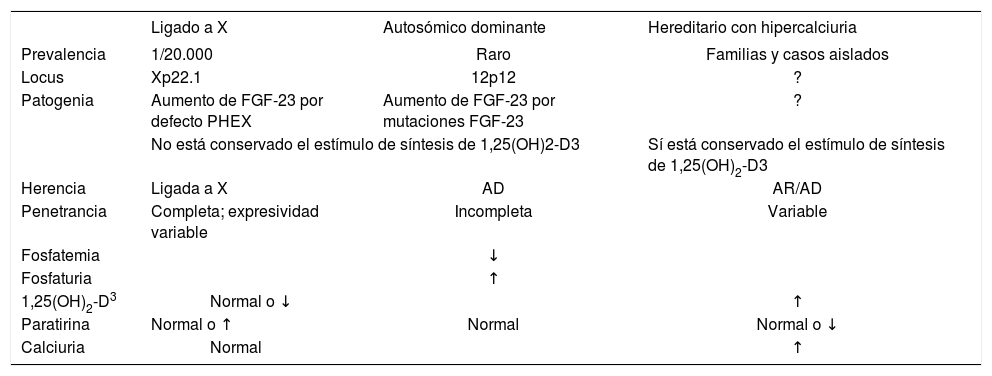
Los tipos de raquitismo hipofosfatémico se recogen en la tabla 48.
Tipos de raquitismo hipofosfatémico
| Ligado a X | Autosómico dominante | Hereditario con hipercalciuria | ||
|---|---|---|---|---|
| Prevalencia | 1/20.000 | Raro | Familias y casos aislados | |
| Locus | Xp22.1 | 12p12 | ? | |
| Patogenia | Aumento de FGF-23 por defecto PHEX | Aumento de FGF-23 por mutaciones FGF-23 | ? | |
| No está conservado el estímulo de síntesis de 1,25(OH)2-D3 | Sí está conservado el estímulo de síntesis de 1,25(OH)2-D3 | |||
| Herencia | Ligada a X | AD | AR/AD | |
| Penetrancia | Completa; expresividad variable | Incompleta | Variable | |
| Fosfatemia | ↓ | |||
| Fosfaturia | ↑ | |||
| 1,25(OH)2-D3 | Normal o ↓ | ↑ | ||
| Paratirina | Normal o ↑ | Normal | Normal o ↓ | |
| Calciuria | Normal | ↑ | ||
1,25(OH)2-D3: 1,25 dihidroxivitamina D3; AD: autosómica dominante; AR: autosómica recesiva; FGF: factor de crecimiento fibroblástico; PHEX: gen que regula el fosfato con semejanza a las endopeptidasas en el cromosoma X.
Es la forma más frecuente de raquitismo. Se transmite siguiendo una herencia dominante ligada al cromosoma X. La mutación se localiza en el gen PHEX (proteína PHEX), lo que condiciona hiperfosfaturia por defecto en la reabsorción proximal de fosfato y, para el grado de hipofosfatemia, disminución de la síntesis de 1,25-(OH)2 vitamina D por una menor actividad de la 1 α-hidroxilasa renal9,10.
Lectura rápida
El estudio inicial debe centrarse en el equilibrio hidrosalino y ácido-base, y el metabolismo fosfocálcico. Para precisar el diagnóstico, pueden ser necesarias pruebas funcionales y determinaciones hormonales.
Principales tubulopatías primariasEl síndrome de Fanconi consiste en una disfunción generalizada del túbulo proximal, que determina aumento de la excreción urinaria de fosfato, bicarbonato, sodio, potasio, aminoácidos, proteínas de bajo peso molecular y glucosa. No se conoce todavía el mecanismo causante, aunque podría deberse a supresión de una fuente importante de energía, a la alteración inespecífica de la permeabilidad celular o a enfermedad en las organelas intracelulares. Lo más frecuente es que se presente en el contexto de otras enfermedades.
Entre las formas de raquitismo hipofosfatémico familiar, el más frecuente es el raquitismo hipofosfatémico ligado al cromosoma X. Se produce hiperfosfaturia por defecto en la reabsorción proximal de fosfato y disminución de la síntesis de 1,25-(OH)2 vitamina D por menor actividad de la 1α-hidroxilasa renal. Es, por tanto, una forma de raquitismo resistente a la vitamina D. Estos pacientes presentan importantes alteraciones óseas y dentarias.
Lectura rápida
Las acidosis tubulares renales pertenecen al grupo de acidosis metabólicas con anion gap plasmático normal o hiperclorémicas. Se clasifican en 3 tipos: a) distal o tipo 1, con alteración de los mecanismos de regulación del equilibrio ácido-base; b) proximal o tipo 2, con alteración en el umbral de reabsorción tubular de bicarbonato, y c) hipercaliémica o tipo 4, en la que se altera el transporte distal de iones hidrógeno dependiente de aldosterona. En el diagnóstico diferencial, es fundamental conocer el potasio sérico y el pH urinario en situación de acidosis.
Se presenta como un raquitismo resistente a la vitamina D, con alteraciones en el crecimiento4, estatura corta inarmónica, deformidades óseas y alteraciones dentarias de la pulpa y la dentina. Radiológicamente, se observa ensanchamiento e incurvación de las metáfisis en los extremos proximal y distal de la tibia y fémur distal11.
En el estudio bioquímico, observamos hipofosfatemia (fósforo: 1,5-2,9mg/dl) con RTP disminuido; normocalcemia; valores de 1,25-(OH)2 vitamina D normales o sólo ligeramente disminuidos; PTH normal o baja y elevación de la fosfatasa alcalina.
El tratamiento consiste en el aporte simultáneo de fosfato oral (1,8-4mg/kg/día) y calcitriol/alfacalcidiol, vigilando signos de intoxicación y la presencia de hipercalciuria (riesgo de nefrocalcinosis). Se valorará el tratamiento con hormona de crecimiento (GH) humana recombinante12. cuando hay deformidades establecidas, pueden realizarse osteotomías correctoras.
Acidosis tubular renalLa ATR es un síndrome clínico de acidosis metabólica causada por un defecto de reabsorción tubular de bicarbonato, o en la excreción urinaria de hidrogeniones. Se trata de una acidosis metabólica hiperclorémica con anion gap plasmático normal ([(Na+) – (Cl– + HCO3–)] = 12 ±2mEq/l)13.
En el diagnóstico diferencial de las ATR (tabla 5)14, es fundamental valorar las cifras de potasio plasmático y el estudio del pH y el anion gap urinario; en ocasiones, es preciso realizar pruebas funcionales. En la ATR proximal se conservan los mecanismos renales de acidificación de la orina.
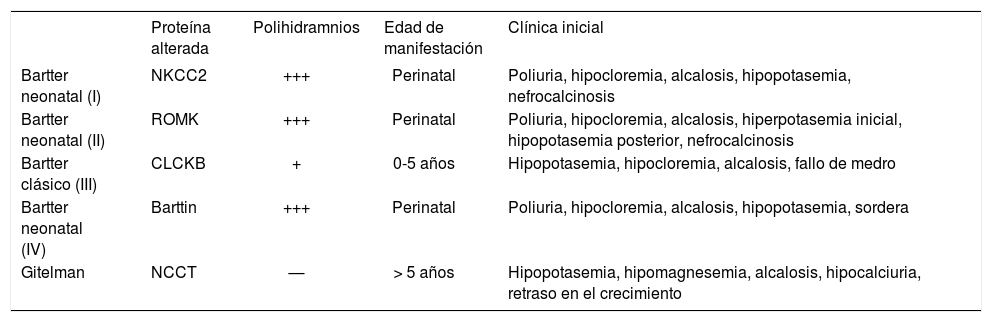
Diagnóstico diferencial entre el síndrome de Bartter y el síndrome de Gitelman
| Proteína alterada | Polihidramnios | Edad de manifestación | Clínica inicial | |
|---|---|---|---|---|
| Bartter neonatal (I) | NKCC2 | +++ | Perinatal | Poliuria, hipocloremia, alcalosis, hipopotasemia, nefrocalcinosis |
| Bartter neonatal (II) | ROMK | +++ | Perinatal | Poliuria, hipocloremia, alcalosis, hiperpotasemia inicial, hipopotasemia posterior, nefrocalcinosis |
| Bartter clásico (III) | CLCKB | + | 0-5 años | Hipopotasemia, hipocloremia, alcalosis, fallo de medro |
| Bartter neonatal (IV) | Barttin | +++ | Perinatal | Poliuria, hipocloremia, alcalosis, hipopotasemia, sordera |
| Gitelman | NCCT | — | > 5 años | Hipopotasemia, hipomagnesemia, alcalosis, hipocalciuria, retraso en el crecimiento |
—: ausente; +: infrecuente; +++: muy frecuente.
El anion gap urinario2 valora de forma indirecta la concentración urinaria de amonio. Se calcula como la diferencia de: (Nao+ + Ko+) – (Clo–) = aniones no cuantificadoso cationes no cuantificadoso (NH4o+). En condiciones normales, es positivo o próximo a 0. Si hay acidosis metabólica, siempre que el mecanismo renal esté conservado, debe aumentar mucho la excreción urinaria de NH4+ y hacerse negativo (paralelamente aumenta la eliminación de cloro para mantener la electronegatividad). Así, el anion gap urinario es negativo en la ATR proximal y positivo en las ATR distal e hipercaliémica. En ocasiones es difícil cuantificarlo por imprecisión de las determinaciones de los iones en orina.
Se pueden clasificar según el esquema siguiente:
- —
ATR proximal (tipo 2)
Alteración de la reabsorción tubular del HCO3– filtrado con reducción de su umbral de eliminación.
- —
ATR distal (tipo 1)
Alteración de la secreción distal de H+:
- a)
De secreción.
- b)
De voltaje.
- c)
De gradiente.
- d)
De aceptores de H+.
- a)
- —
ATR mixta (tipo 3)
ATR distal asociada a inmadurez tubular proximal transitoria en lactantes.
- —
ATR hipercaliémica (tipo 4)
Alteración de la amoniogenia asociada a situación de hipoaldosteronismo e hiperpotasemia.
Se produce por alteración de la secreción de iones hidrógeno en el túbulo distal. Hay incapacidad renal para eliminar el exceso de ácido: el pH urinario no disminuye por debajo de 5,5 en situación espontánea de acidosis plasmática (ATR distal completa) o bien tras sobrecarga ácida (ATR distal incompleta).
Se manifiesta clínicamente con un cuadro general caracterizado por fallo de medro, anorexia, vómitos y debilidad muscular de intensidad variable. Dado que el hueso se comporta como principal tampón del ácido plasmático, la situación de acidosis crónica obstaculiza el crecimiento y el desarrollo esquelético. Además, la acidosis inhibe la secreción de GH (reversible). El pronóstico lo determina la tendencia a nefrocalcinosis temprana (hipercalciuria e hipocitraturia asociadas) con posible evolución a la insuficiencia renal. El tratamiento temprano previene su aparición.
- 1.
Primaria:
- •
Esporádica.
- •
Hereditaria:
- a)
Clásica:
- —
Herencia autosómica dominante (AD) (mutación en el gen SLC4A1 en 17q 21–22 que codifica el cotransportador Cl–/HCO3– [proteína AE1]).
- —
AR aislada (mutaciones en el gen ATP6V0A4 en 7q33-34 que codifica la subunidad α4 del transportador H+–K+–ATPasa).
- —
- b)
Asociada a sordera neurosensorial.
- —
AR (ATP6V1B1 en 2p13 que codifica la subunidad B1 de la H+−ATPasa).
- —
- a)
- •
- 2.
Secundaria: nefrocalcinosis, enfermedades genéticas y autoinmunitarias, tóxicos.
Se produce por alteración de la reabsorción tubular del HCO3–filtrado con la disminución de su umbral de excreción (en condiciones normales cuando excede 22–26mEq/l, en lactante y adulto, respectivamente). Normalmente, la concentración en plasma se mantiene entre 14 y 20mEq/l, sin producir acidosis más graves porque los mecanismos compensatorios distales están conservados. Puede manifestarse aisladamente o bien en el contexto de un síndrome de Fanconi de cualquier etiología, que es lo más frecuente. Las manifestaciones clínicas se reducen al retraso del crecimiento y vómitos en lactantes.
- 1.
Primaria:
- •
Esporádica: transitoria (lactantes).
- •
Hereditaria: persistente.
- a)
AR asociada a anomalías oculares y retraso mental (mutación en el gen SLC4A4 en 4p21 que codifica el transportador NBC-1).
- b)
AD.
- •
- 2.
Secundaria: síndrome de Fanconi, hiperparatiroidismo, enfermedad quística renal, cardiopatía cianótica, etc.
Lectura rápida
En la hipercalciuria idiopática, se observa excreción elevada de calcio en orina sin hipercalcemia ni otras causas conocidas de hipercalciuria. Es la primera causa de urolitiasis en la infancia. En la patogenia confluyen un incremento de la absorción intestinal de calcio y de la resorción ósea y el descenso de su reabsorción tubular. Además de las complicaciones de litiasis y nefrocalcinosis, también se ha observado un descenso de masa ósea en estos pacientes.
Debe hacerse diagnóstico diferencial con causas que ocasionen pérdidas digestivas de bicarbonato.
Lectura rápida
El síndrome de Bartter es un trastorno en el que se encuentra alterada la reabsorción de sal en el asa de Henle. Se caracteriza por alcalosis metabólica con hipocaliemia, hiperaldosteronismo (hiperactivación del sistema reninaangiotensina-aldosterona e hiperplasia del aparato yuxtaglomerular) e hiperprostaglandinismo con presión arterial normal. Se diferencian 2 fenotipos: la forma neonatal (tipos I, II y IV) y la forma clásica (tipo III). El pronóstico depende del desarrollo de una nefropatía tubulointersticial crónica en la evolución; es peor en los de inicio neonatal, por el riesgo añadido de desarrollar nefrocalcinosis.
Se produce por un defecto en la secreción distal de H+ y K+ por deficiencia o resistencia a la aldosterona (hipoaldosteronismo/seudohipoaldosteronismo) y por defecto en la amoniogenia causada por la propia hiperpotasemia.
- 1.
Primaria.
Seudohipoaldosteronismo tipo I (AD, forma renal, y AR, forma múltiple) y tipo II o síndrome de Gordon-”shunt del cloro” (AD).
Hipercaliemia transitoria del lactante (esporádica).
- 2.
Secundaria.
Deficiencia de aldosterona (suprarrenal).
Hipoaldosteronismo hiporreninémico en enfermedad renal crónica.
Hipoaldosteronismo hiporreninémico en glomerulonefritis aguda.
Seudohipoaldosteronismo tipo I secundario a enfermedad obstructiva o infecciosa (PNA).
Drogas o tóxicos (antiinflamatorios no esteroideos, trimetoprima, captopril, ciclosporina, heparina).
En el tratamiento de la ATR proximal se requieren dosis altas de bicarbonato (hasta 10–20mEq/kg/día) y, en ocasiones, es necesario asociar diuréticos tiazídicos para aumentar su reabsorción. En la ATR distal, se debe administrar bicarbonato o citrato potásico en dosis de 3–5mEq/kg/día y se controlará la calciuria. En la ATR tipo IV, si es posible, debe realizarse tratamiento etiológico, tratamiento de la hiperpotasemia y, si hay hipoaldosteronismo, puede administrarse fluorhidrocortisona.
Hipercalciuria idiopáticaSe define por una excreción elevada de calcio en orina sin hipercalcemia ni otras causas conocidas de hipercalciuria. Es la primera causa de urolitiasis en la infancia15. Parece que hay agrupación familiar, aunque con dudas acerca del patrón de herencia. En la patogenia intervienen mecanismos que incrementan la absorción intestinal de calcio y la resorción ósea y factores que disminuyen su reabsorción tubular.
Las manifestaciones clínicas más frecuentes son hematuria macroscópica y/o microscópica, disuria, síndrome de polaquiuria-urgencia miccional, incontinencia urinaria, enuresis, dolor abdominal, orina turbia matutina e infección urinaria. Como complicaciones frecuentes, urolitiasis y nefrocalcinosis.
Se ha observado que estos niños pueden presentar retraso del crecimiento. La reducción de la densidad mineral ósea que se observa en pacientes hipercalciúricos, en probable relación con el estímulo de la resorción ósea, podría estar implicada16–18.
Además del estudio del metabolismo fosfocálcico, se debe realizar análisis en plasma y de las excreciones en orina de sodio, potasio, cloro y magnesio. La determinación de citrato y oxalato en orina de 24h permite valorar el riesgo de litiasis. Se realizará ecografía renal para determinar posibles imágenes de cálculos y/o nefrocalcinosis y la densitometría ósea hacia los 12 años de edad.
Tratamiento- —
Dieta pobre en sodio (limita la excreción) y rica en potasio (facilita la reabsorción); que no supere los requerimientos diarios de proteínas19; controlada en calcio y oxalato20.
- —
En algunos casos, será necesario administrar citrato potásico y/o tiacida (vigilar citraturia, potasio plasmático, lipidograma)21.
- —
Líquidos abundantes.
Otros trastornos tubulares que cursan con hipercalciuria son:
- —
Síndrome de Fanconi.
- —
Enfermedad de Dent.
- —
Acidosis tubular renal distal.
- —
Raquitismo hipofosfatémico con hipercalciuria.
- —
Síndrome de Bartter.
- —
Hipomagnesemia con hipercalciuria y nefrocalcinosis.
El síndrome de Bartter es un trastorno hereditario (AR) en el que se encuentra alterada la reabsorción de sal en el asa de Henle, con alteraciones muy similares a las que tienen lugar en los pacientes tratados con diuréticos tipo furosemida22.
El asa de Henle se encarga de la reabsorción del 45% de sodio, cloro y potasio filtrados, del 25% del agua y de cantidades significativas de calcio y magnesio1.
La pérdida salina condiciona una situación compensatoria de hiperaldosteronismo (hiperactivación del sistema renina-angiotensina-aldosterona e hiperplasia del aparato yuxtaglomerular), a pesar de lo cual la presión arterial se mantiene normal (disminución del volumen intravascular, aumento de la síntesis de prostaglandinas y otras sustancias vasodilatadoras)23. Se diferencian 2 fenotipos: la forma neonatal (tipo I: mutación en el gen SLC12A1 [15q 15–21] que codifica el cotransportador Na+− K+−2Cl− (BSC1 o NKCC2); tipo II: mutación en el gen KCNJ1 (11q 24–25) que codifica el canal de potasio que recicla el potasio hacia la luz tubular (ROMK); tipo IV: mutación en el gen BSND (1p31) que codifica la proteína Barttin, subunidad β de los canales de cloro ClC-Ka y Kb), y la forma clásica (tipo III: mutación en el gen CLCNKB [1p36] que codifica el canal de cloro ClC-Kb)24.
La forma clásica (de inicio en los primeros 2 años de vida) se caracteriza por presentar poliuria y polidipsia, avidez por la sal y, en contexto de la depleción crónica de volumen, alcalosis metabólica hipoclorémica e hipopotasemia (lo más característico)25, normocalciuria o hipercalciuria y natremia normal o levemente disminuida. Estos niños presentan anorexia, vómitos, estreñimiento, episodios repetidos de deshidratación con retraso del desarrollo ponderoestatural e intelectual. Más tardíamente, astenia, debilidad muscular, calambres y tetanias. El pronóstico depende del desarrollo de una nefropatía tubulointersticial crónica por la acción fibrogénica de la angiotensina II y la hipopotasemia crónica, con posible alteración del FG en la evolución26,27.
La forma neonatal se inicia como parto prematuro con antecedente de polihidramnios, poliuria marcada de difícil control e hipercalciuria con nefrocalcinosis temprana. Algunos pacientes presentan un fenotipo peculiar; puede asociarse a sordera neurosensorial (tipo IV)28. El síndrome de Bartter neonatal tipo II puede presentarse al inicio con acidosis metabólica, hiponatremia e hiperpotasemia29. En los casos neonatales, el riesgo de disfunción renal es mayor por el desarrollo de nefrocalcinosis.
Debe realizarse diagnóstico diferencial con el síndrome de Gitelman, de presentación generalmente en la preadolescencia o en la edad adulta y de curso clínico benigno (tabla 5)30.
El tratamiento se basa en la administración de suplementos de potasio y de inhibidores de las prostaglandinas (indometacina, tolmetina sódica) que limitan las pérdidas de agua y electrolitos por reducción del FG. Algunos casos se pueden beneficiar del empleo de espironolactona. Pueden precisar aportes de magnesio.
Diabetes insípida nefrogénicaLa diabetes insípida nefrogénica es una enfermedad caracterizada por la resistencia renal al efecto de la hormona antidiurética (ADH), con la consecuente incapacidad para concentrar la orina a pesar de valores normales o elevados de hormona circulante31,32.
- 1.
Primaria: hereditaria33,34.
- —
Ligada al cromosoma X (90%): mutación en el gen AVPR2 (Xq28) que codifica el receptor V2 de la vasopresina en el túbulo colector. Las mujeres presentan formas leves de la enfermedad.
- —
AR (10%) y AD (1%): mutación en el gen AQP2 (12q13) que codifica el canal de agua dependiente de vasopresina (aquaporina 2).
- —
- 2.
Secundaria: adquirida. Enfermedad renal con afectación túbulo-intersticial, enfermedades sistémicas, fármacos (sales de litio, demeclociclina)35 y otros36.
Lectura rápida
La diabetes insípida nefrogénica es una enfermedad caracterizada por la resistencia renal al efecto de la hormona antidiurética con incapacidad para concentrar la orina, a pesar de valores normales o elevados de hormona circulante. Se presenta con poliuria, hipostenuria y polidipsia secundaria, con episodios repetidos de deshidratación hipernatrémica muy graves, trastornos en la alimentación, vómitos, estreñimiento y escasa ganancia ponderal. La poliuria puede generar megavejiga, megauréter e hidronefrosis. Puede aparecer insuficiencia renal crónica por trombosis renal en los episodios de deshidratación.

Se manifiesta con poliuria (hasta 5ml/kg/h), hipostenuria (osmolalidad: 50–100mOsm/kg; densidad < 1.010) y polidipsia secundaria, con episodios repetidos de deshidratación hipernatrémica muy graves (causantes del retraso mental secundario). Además, fiebre sin foco e irritabilidad; trastornos en la alimentación, vómitos, estreñimiento y escasa ganancia ponderal. La poliuria determina en ocasiones megavejiga, megauréter e hidronefrosis. Puede aparecer insuficiencia renal crónica consecuencia del daño parenquimatoso por trombosis renal en los episodios de deshidratación. El diagnóstico se retrasa en ocasiones hasta los 3–4 años por lo inespecífico de los síntomas en etapas tempranas37.
La sospecha diagnóstica se basa en la sintomatología descrita, junto con osmolalidad en sangre > 300mOsm/kg y en orina < 200mOsm/kg, con hipernatremia (sodio plasmático > 147mmol/l) e hipercloremia, elevación de creatinina y urea de causa prerrenal y acidosis según el grado de deshidratación, con FG normal.
Para establecer el diagnóstico debe comprobarse la ausencia de respuesta ante valores elevados de ADH plasmática, bien mediante el test de restricción hídrica prolongada, bien después de la administración del análogo DDAVP. Si la osmolaridad urinaria en ayunas tras la restricción hídrica nocturna es > 800 mosm/kg, consideramos que la capacidad de concentración es normal: probable potomanía. El tratamiento es sintomático con reposición de las pérdidas urinarias y asegurar un adecuado aporte calórico, para lo cual, en ocasiones, se requiere mantener nutrición enteral continua (u ofrecer agua cada 2h día y noche hasta que el niño tenga libre acceso al agua). Se debe disminuir la ingesta de solutos (sodio 0,7-1mEq/kg/día y proteínas 2g/kg/día) para evitar la carga renal osmolar38. Pueden emplearse diuréticos tiacídicos (asociados a amilorida) e inhibidores de las prostaglandinas para disminuir la poliuria39–41.


