




Introducción. El neurofibroma solitario es una lesión benigna que se origina de los nervios. Se presenta en adultos jóvenes sin predominio de sexo. La escisión es considerada el tratamiento de elección. Caso clínico. Mujer de 37 años, remitida a nuestro servicio por edema en el pie y la pierna derecha de varios meses de evolución. En la exploración clínica se aprecia tumoración sólida, rodadera, no adherida a planos profundos, de gran tamaño, ocupando la fosa poplítea. La exploración con eco-Doppler aprecia permeabilidad de arteria y vena poplíteas, muy desplazadas de su eje. La angio-RM confirma una formación quística en hueco poplíteo, de 60mm × 40mm. La sintomatología de la paciente y el riesgo de complicaciones (trombosis venosa, daño a la arteria poplítea por traumatismo crónico) determinaron la indicación quirúrgica. En la intervención se identifica una tumoración sólida y encapsulada, sin conexión con paquete vascular, pero íntimamente dependiente de trayecto nervioso ciático poplíteo externo. Se extirpó en su totalidad. El postoperatorio transcurrió con normalidad. La paciente permanece asintomática y en las sucesivas revisiones no se ha objetivado recidiva ni complicaciones vasculares. Conclusión. Los neurofibromas son tumores benignos que se localizan en cualquier lugar del organismo y pueden alcanzar gran tamaño. Deben ser extirpados quirúrgicamente si provocan síntomas o complicaciones (dolor, compresión, deterioro neurológico) y deben ser estudiados ante sospecha de degeneración maligna. El pronóstico tras resección quirúrgica es bueno y la recurrencia es rara. [ANGIOLOGÍA 2009; 61: 359-64]
Introduction. The solitary neurofibroma is a benign lesión that originates from the peripheral nerves. It presents in young adults of either sex. Excision is considered as the treatment of choice. Clinical case. A 37 year-old woman referred to our department due to an oedema that had developed over several months in the right foot and leg. A large, solid, rolling tumour, which was non-adherent to the deep planes, was seen occupying the popliteal fossa. The echo-Doppler examination showed the patency of the popliteal artery and vein, which were much displaced form their axis. The Angio-MRI confirmed a cystic formation of 60mm × 40mm in the popliteal fossa. Surgery was indicated, due to the patient symptoms and the risk of complications (venous thrombosis, damage to the popliteal artery due to chronic trauma). During surgery, a solid encapsulated tumour was identified, which was disconnected from the vascular bundle, but closely dependent on the external popliteal sciatic nerve trajectory. It was completely removed. There were no post-operative complications. The patient remains asymptomatic and there has been no recurrence or vascular complications in successive reviews. Conclusion. Neurofibromas are benign tumours that can be found in any part of the body and can be very large. They must be surgically removed if there are symptoms or complications (pain, compression, neurological deterioration) and they must be study if malignant degeneration is suspected. The prognosis after surgical resection is good and recurrence is rare. [ANGIOLOGIA 2009; 61: 359-64]
Los neurofibromas pueden presentarse como lesiones solitarias o asociadas a una neurofibromatosis (neurofibromas cutáneos y extracutáneos múltiples, manchas café con leche, hamartomas del iris, etc.). La tríada clínica clásica de la neurofibromatosis es: lesiones cutáneas, deformidades esqueléticas y deficiencia mental.
Los neurofibromas se originan de los nervios y pueden presentarse en cualquier lugar donde se encuentren terminaciones nerviosas. Pueden ser proliferaciones de las células de Schwann, células perineurales y fibroblastos endoneurales, por lo que se puede decir que son proliferaciones policlonales y pueden considerarse como hiperplasias de todos los elementos neurales [1].
Entre todos los tipos de tumores neurogénicos, existen dos tipos principales de tumores benignos de fibras nerviosas: el neurofibroma y el schwannoma. La diferenciación entre estas dos tumoraciones es de tipo histológico, en el schwannoma las células se encuentran mayormente diferenciadas, además las lesiones son encapsuladas y no contienen axones.
Los tipos de neurofibroma son: localizado, múltiple, difuso y plexiforme.
La forma localizada es la más común (90% de los casos). La gran mayoría son solitarios y no asociados a neurofibromatosis o enfermedad de Von Recklinghausen.
La forma difusa afecta a niños y adultos jóvenes. Compromete el tejido subcutáneo de cabeza y cuello. Generalmente son lesiones aisladas y no asociadas a neurofibromatosis.
La forma plexiforme se considera patognomónica de neurofibromatosis tipo I. Representa el compromiso difuso de un tronco neural y sus ramas. El desarrollo de este tipo de lesiones ocurre principalmente durante la niñez y precede la aparición de neurofibromas cutáneos [2].
El neurofibroma solitario es una lesión benigna, de crecimiento lento que generalmente se manifiesta en los adultos. Clínicamente se pueden describir como neoformaciones del color de la piel, salientes o pediculadas, de consistencia blanda, y se ha descrito que pueden ser muy pruriginosos. Representan el 90% de los neurofibromas y generalmente se presentan entre los 20 y los 30 años de edad sin predominio de sexo. La escisión es considerada el tratamiento de elección en estos casos. Los neurofibromas clásicos son tumores bien circunscritos, no encapsulados, localizados en la dermis y algunas veces se extienden hasta el tejido celular subcutáneo superficial, están compuestos por células que se describen como fusiformes, citoplasma eosinófilo y núcleo oval basófilo, además de colágeno y mucina.
Caso clínicoMujer de 37 años de edad remitida a nuestro servicio por edema en pie y pierna derecha de meses de evolución. Refiere notarse desde hace 2 años una tumoración en hueco poplíteo, que ha crecido y que en los últimos meses le produce molestias.
Como antecedentes de interés destacan: tabaquismo y dos embarazos. Intervenida de tumoración en pantorrilla izquierda 4 años antes, pero desconoce su etiología o datos histológicos.
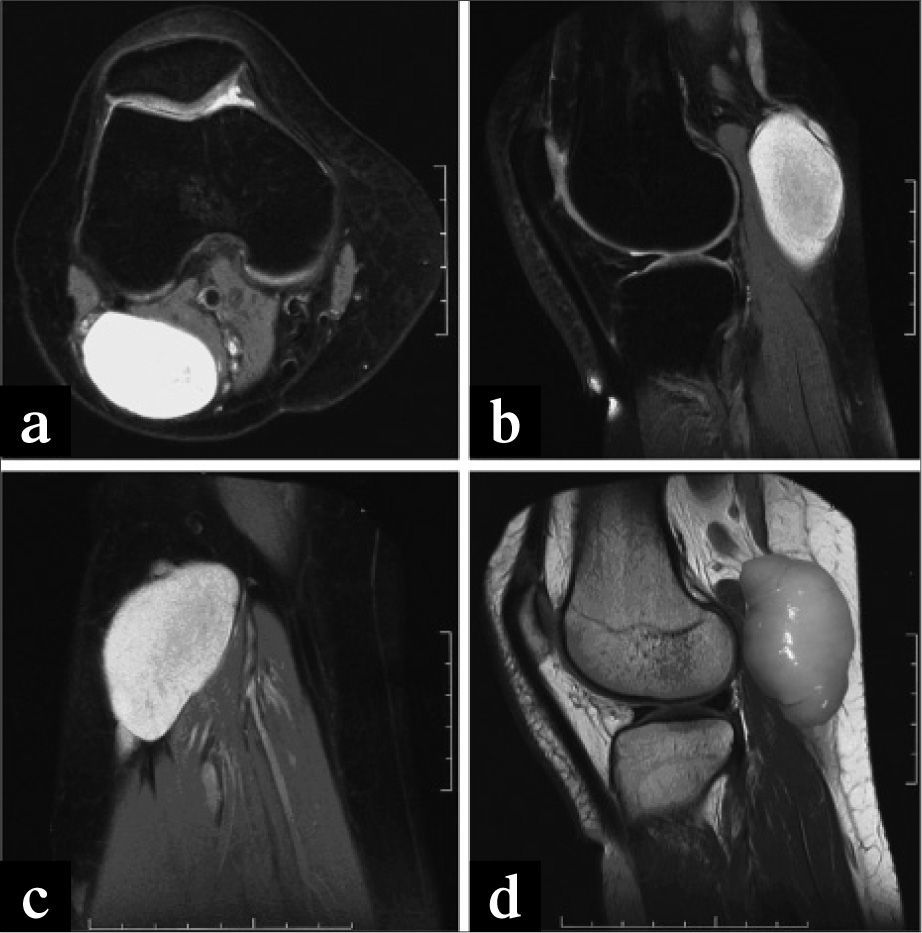
En la exploración se aprecia tumoración sólida (Fig. 1a), rodadera, de gran tamaño, sin adherencia a planos profundos, ocupando completamente la fosa poplítea. Pulsos arteriales normales a todos los niveles.
 Visión lateral de la tumoración poplítea. b) Incisión en jarretera. Se observa el nervio ciático poplíteo externo (*).")
La exploración con eco-Doppler revela permeabilidad de vena safena externa y de arteria y vena poplíteas, pero muy desplazadas de su eje, y una tumoración sólida en íntimo contacto con la vena poplítea que no cumplía criterios de quiste de Baker y en la que no existía una conexión clara con la cavidad articular, por lo que se decidió realizar una angio-RM.
En la angio-RM se detecta una formación de localización posteroexterna en hueco poplíteo, de paredes lisas, sin conexión con la cápsula articular, desplazando el eje vascular y de dimensiones de 60mm × 40mm (Fig. 2). En la anamnesis y exploración completa no aparecía indicio de neurofibromas en otras partes del cuerpo.
 Imágenes del neurofibroma en la ARM. d) Imagen del neurofibroma extirpado superpuesta sobre la imagen de ARM.")
La sintomatología de la paciente y el riesgo de complicaciones por el desplazamiento del eje vascular arteriovenoso y por la ocupación del hueco poplíteo (trombosis de vena poplítea, trombosis de vena safena externa, daño arterial por trauma crónico) determinaron la indicación quirúrgica.
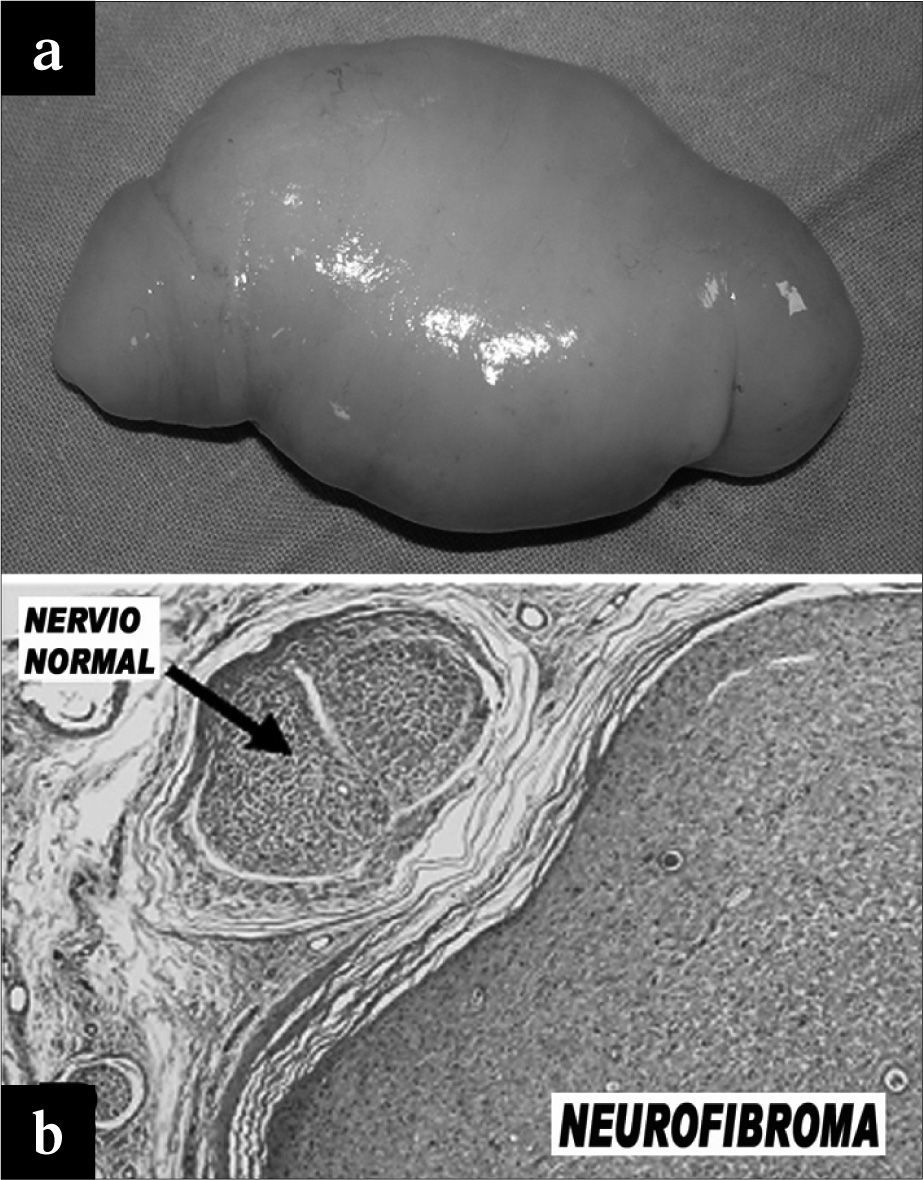
Bajo anestesia general y en posición de decúbito prono (Fig. 1b) se realizó un abordaje posterior e incisión en "S itálica" en hueco poplíteo. Al abrir la fascia, la tumoración sólida se hernió espontáneamente (Fig. 3a). No tenía conexión con el paquete vascular, pero dependía íntimamente del trayecto nervioso ciático poplíteo externo. Finalmente se extirpó en su totalidad.
 Neurofibroma tras su extirpación. b) Histología.")
El examen anatomopatológico y microscópico evidenció una tumoración bien delimitada y no encapsulada, constituida por células fusiformes que en algunas zonas exhibían pleomorfismo e hipercromía e incluso seudoinclusiones, disponiéndose en patrón fascicular y laxo con material proteico entre los fascículos y células inflamatorias entremezcladas, algunas con aspecto de mastocitos (Fig. 3b). No se observaron mitosis, células atípicas, signos invasivos, necrosis ni focos de hemorragia. Estos hallazgos eran compatibles con el diagnóstico de neurofibroma.
El postoperatorio transcurrió con normalidad sin complicaciones derivadas del procedimiento. La paciente se encuentra asintomática y en las sucesivas revisiones clínicas y control mediante eco-Doppler, no se ha objetivado recidiva.
DiscusiónLa mayoría de los neurofibromas son masas pequeñas y solitarias que raramente alcanzan gran tamaño. Normalmente se manifiestan clínicamente como lesiones blandas de la piel, que pueden ser pruriginosas. A pesar de que la mayoría de los neurofibromas son solitarios, debe considerarse siempre la posible asociación con neurofibromatosis.
El compromiso ocasionado por los neurofibromas depende en buena parte de su localización: las lesiones cutáneas generalmente causan deformidad, mientras que las lesiones más profundas tienden a generar compromiso funcional. A pesar de ser benignos en su mayoría, algunos causan destrucción secundaria por la presión ejercida, por lo que los síntomas van a depender del tamaño de la lesión. Es importante tener en cuenta que la aparición de dolor o crecimiento rápido de la masa puede sugerir una transformación maligna.
La clínica varía según la localización, el tamaño, la relación con los órganos vecinos, el desplazamiento y la presión ejercida sobre las estructuras cercanas, que van a generar compromiso de la funcionalidad de éstas, manifestándose clínicamente de forma específica en cada caso, por lo que los motivos de consulta pueden variar ampliamente.
Por esa razón, se insiste en una adecuada anamnesis –tiempo de evolución, crecimiento de la masa, presencia de dolor, prurito u otra sintomatología asociada– y en un examen físico riguroso –palpación detallada para determinar la forma, la consistencia, la movilidad o descartar presencia de tumores en otras localizaciones– [3].
En nuestro caso el síntoma principal de la paciente eran las molestias y el edema del pie y de la pierna derecha causados por una masa localizada en el hueco poplíteo de crecimiento lento asociado al compromiso vascular de retorno venoso provocado por la desviación manifiesta del eje vascular y la ocupación de espacio, aunque la vena poplítea estaba permeable.
A veces se dificulta la sospecha clínica y el diagnóstico de neurofibroma por su presentación en una localización inusual. Las presentaciones infrecuentes de neurofibroma solitario en pacientes sin neurofibromatosis que hemos encontrado en la literatura consultada son: sistema respiratorio (cavidad nasal, tonsila palatina, subglótico), intramedular, sistema digestivo (paladar blando, íleon, colon, conducto biliar común, canal anal) y sistema urogenital (riñón, cordón espermático, escrotal, vulvar).
De similar localización al caso que presentamos, se ha encontrado la descripción de un caso de neurofibroma en una mujer de 45 años que presentaba una tumoración gigante ulcerada en hueco poplíteo [4], una referencia a los hallazgos ultrasonográficos de un neurofibroma plexiforme de fosa poplítea [5], y un caso de un neurofibroma plexiforme gigante que englobaba el nervio peroneo común [6].
En nuestro caso la primera sospecha diagnóstica por la localización fue de quiste de Baker –con el que debe hacerse diagnóstico diferencial–, pero los estudios de imagen lo descartaron.
Todos los tumores neurogénicos tienen hallazgos clínicos y radiológicos similares [7]. En general, se manifiestan en estudios de imagen como bien definidos, blandos y lobulados. Se pueden observar calcificaciones en cualquiera de estos tumores, por lo que no es un dato útil para orientar el diagnóstico. En ocasiones es difícil distinguir un neurofibroma con diferentes grados de atipia de un tumor maligno. Lo que puede sugerir malignidad es observar focos metastáticos distantes. Se considera que la valoración microscópica y los estudios inmunohistoquímicos son esenciales para el diagnóstico exacto, ya que en algunos casos son requeridos para distinguir los neurofibromas de otros tumores [8].
Los neurofibromas son tumores benignos, que generalmente no llegan a desarrollar transformación maligna. Sin embargo, se asocian a neurofibromatosis en un 2-5% de los casos, especialmente en la presentación plexiforme [9].
El tratamiento para el neurofibroma solitario es la resección quirúrgica completa de este, en lo posible debe extraerse completo, en especial si el riesgo de recurrencia es considerado alto, ya que otras medidas terapéuticas no resultan efectivas. La técnica quirúrgica y las formas de abordaje para el tratamiento varían según su localización y el compromiso que esté generando. Por tanto, puede ser necesario un manejo interdisciplinario entre diferentes especialidades quirúrgicas en muchos casos, sobre todo en localizaciones especiales, porque tras un estudio detallado y un análisis individualizado de cada caso se puede definir el mejor abordaje quirúrgico. El pronóstico tras resección quirúrgica es bueno y la recurrencia es rara.
Cabe concluir que los neurofibromas son tumores benignos que pueden localizarse en cualquier lugar del organismo. Deben ser extirpados quirúrgicamente si provocan síntomas o complicaciones, y deben ser estudiados anatomopatológicamente para descartar su malignidad.
