


La sepsis es una enfermedad de origen infeccioso con una prevalencia de 2.911 por 100.000 pacientes en Estados Unidos, y se caracteriza por el desbalance de la respuesta pro y antiinflamatoria por parte del sistema inmune. Sin embargo, la falta de comprensión de la respuesta del huésped frente a la infección justifica la importancia de su debate. En esta revisión no sistemática se plantea la relación funcional que existiría entre el receptor de adenosina A2A (A2A), óxido nítrico (NO) y factor de crecimiento vascular (VEGF), los cuales regulan la vasodilatación y la permeabilidad microvascular, y que en la sepsis se encuentran alterados. La evidencia revisada muestra que en el medio extracelular expuesto a infección se genera un estado de hipoxia celular, la que aumenta la producción de adenosina, nucleósido derivado del metabolismo del ATP. Esta molécula, al activar el receptor A2A presente en células del sistema inmune y endoteliales es capaz de reducir la respuesta inmunológica y aumentar la permeabilidad vascular, lo que a su vez ha sido asociado con una mayor progresión bacteriana y fallo multisistémico. Molecularmente, A2A activa la síntesis y producción de NO y VEGF. Por su parte, VEGF, al activar su receptor tipo 2 aumenta también la producción de NO, generando un circulo potenciador del efecto inmunológico y vascular. Pese a estas evidencias no hemos encontrado estudios que relacionen directamente estos 3 actores, A2A-NO y VEGF en la sepsis. Por ello, a la luz de la evidencia revisada, proponemos que la vía A2A-NO-VEGF sería un blanco de estudio en la fisiopatología y tratamiento de la sepsis.
Sepsis is an infectious disease with a prevalence of 2.9 per 100 individuals in the United States. It is characterised by an unbalanced pro- and anti-inflammatory response driven by the immune system. However, further discussion is needed due to the lack of understanding on the details of the host response to the infection. This non-systemic review addresses the functional relationship between the Adenosine 2A receptor (A2A), Nitric Oxide (NO), and Vascular Epithelial Growth Factor (VEGF), which regulate vasodilation and micro-vascular permeability, which in turn are affected in sepsis. The evidence reviewed shows that an state of cellular hypoxia is generated due to infection, which increases the production of adenosine (ADO), a nucleoside derived from the metabolism of ATP. This molecule, by activating A2A receptor present in cells of the immune and endothelial systems reduces the immune response and increasing vascular permeability, which in turn has been associated with greater bacterial progression and multisystem failure. Molecularly, A2A activates the synthesis and production of NO and VEGF. In this regard, VEGF, by activating its type 2 receptor (VEGFR2) also increases NO production. This generates a cycle that enhances the immunological and vascular effect. Despite this, no studies were found that directly links these three actors, A2A-NO and VEGF, in sepsis. Therefore, taking into account the reviewed evidence, it is proposed that the A2A-NO-VEGF pathway could be a target of study in the pathophysiology and treatment of sepsis.
La definición más difundida de sepsis indica que es un estado patológico caracterizado por una respuesta inflamatoria exacerbada del huésped frente a un patógeno. Sin embargo, actualmente se entiende este cuadro como una respuesta que involucra alteraciones sistémicas que van más allá del sistema inmune, incluyendo los sistemas nervioso, endocrino, de coagulación y vascular1.
El impacto de esta enfermedad en términos de morbimortalidad, así como económicos, es enorme. Específicamente, en un estudio de cohorte realizado en pacientes del sistema hospitalario de Estados Unidos se estimó una prevalencia de sepsis de 2.911 por 100.000 usuarios. Además, la mortalidad atribuible a la sepsis varió entre 20-80% de acuerdo a la severidad del cuadro y a las condiciones basales del paciente. En lo económico el coste promedio por caso fue de 22.100 dólares americanos, con costes anuales totales de 16,7 billones2.
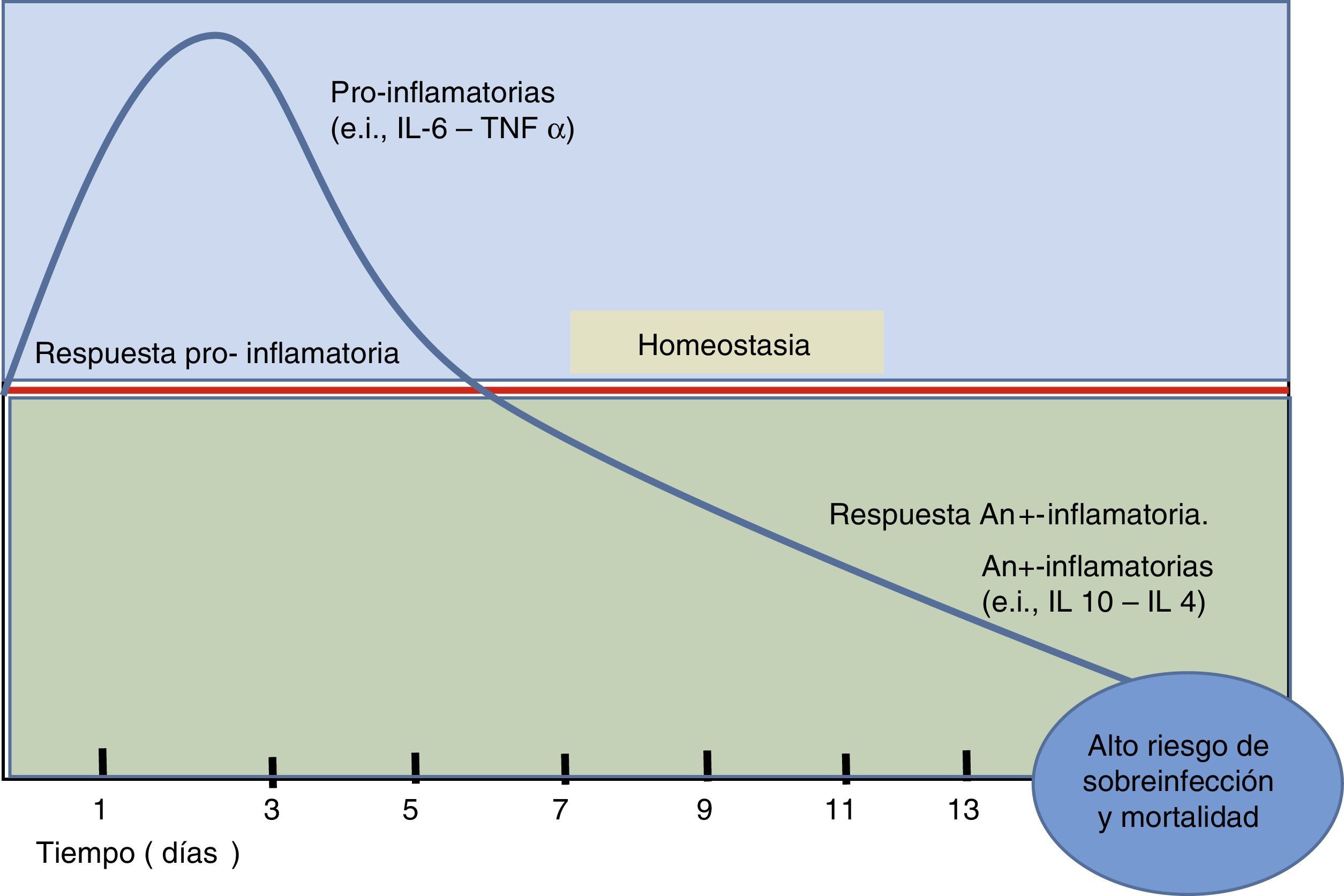
En la sepsis ocurre activación del sistema inmune, caracterizado por aumento de la síntesis de interleucinas pro y antiinflamatorias (fig. 1). Este incremento, sin embargo, no es un estado estático, sino más bien dinámico. Particularmente, en el estado de sepsis se puede evidenciar un desequilibrio entre la síntesis de las interleucinas proinflamatorias (como interleucina 1 [IL-1] o IL-6; o factor de necrosis tumoral alfa [TNF-α]) y antiinflamatorias (como IL-10 e IL-4); a favor de estas últimas lleva a un estado de inmunosupresión o «inmunoplejía», lo cual evita que el organismo pueda defenderse adecuadamente de los agentes invasores. Esta incrementada respuesta antiinflamatoria en la sepsis está asociada con un aumento de la carga bacteriana y complicaciones macrohemodinámicas y de la microcirculación, lo cual finalmente lleva a poner en riesgo la vida del paciente.

Balance de interleucinas pro y antiinflamatorias durante la sepsis. La figura esquematiza el disbalance entre interleucinas pro y antiinflamatorias en sepsis. Esto genera un estado de inmunosupresión que permitiría la progresión bacteriana y subsecuentemente la muerte del individuo. La línea roja significa el estado normal o de hemostasia. Se grafica el tiempo en días.
La sepsis se asocia también a un estado conocido como «disfunción endotelial», en la cual esta célula pierde la capacidad de síntesis de sustancias vasoactivas ligadas a la regulación de la coagulación, del tono vasomotor, la formación de nuevos vasos sanguíneos y la homeostasis vascular en general. Como consecuencia de este rol la disfunción endotelial es un acontecimiento sistémico que se traduce como una alteración de la vasorregulación, desbalance de la liberación de factores pro y antiinflamatorios y daño hipóxico/isquémico. Este estado de disfunción del endotelio se ha asociado con sepsis, vasculitis autoinmunes, estrés oxidativo y estrés nitrosativo, entre otros.
Molecularmente se conoce que los estados de estrés celular, como la inflamación y la hipoxia, asociados a la sepsis, aumentan la producción extracelular del nucleósido adenosina (ADO)3. La adenosina, un nucleósido derivado del metabolismo del ATP, es capaz de activar a una familia de receptores de membrana asociados a proteína G (A1, A2A, A2B y A3). Esta activación de receptores de adenosina regula también múltiples fenómenos fisiológicos, incluyendo regulación del tono vascular, hemostasia y coagulación y angiogénesis, entre otros4. Estos procesos los cumple, al menos en parte, a través de la activación de la síntesis de óxido nítrico (NO) y del factor de crecimiento de endotelio vascular (VEGF). Múltiples grupos5,6 incluyendo el nuestro7,8 han mostrado una interacción unidireccional entre los receptores A2A, NO sintasa endotelial (eNOS), síntesis de NO y expresión de VEGF en condiciones fisiológicas o patológicas. Pero no se conoce la participación de esta vía A2A-NO-VEGF en la sepsis. Sin embargo, se conoce que la elevación de interleucinas proinflamatorias, el estado de hipoxia y de estrés oxidativo incrementan la síntesis y liberación de ADO3, asociado a expresión de la isoforma inducible de NOS (iNOS)9. La iNOS, por su parte, libera NO en cantidades 1.000 veces mayor que la eNOS. Además, en este estado de estrés celular se incrementa también la síntesis de VEGF. El escenario se complica cuando todos estos actores además se autorregulan entre sí.
No se ha estudiado en profundidad cuál sería la relación del aumento de ADO y la vía de señalización A2A-NO-VEGF con el daño vascular de sepsis. Con ello el objetivo de esta revisión es proponer un mecanismo en el cual se integre la vía A2A-NO-VEGF con el daño vascular secundario a la desregulación de la respuesta inmune en sepsis.
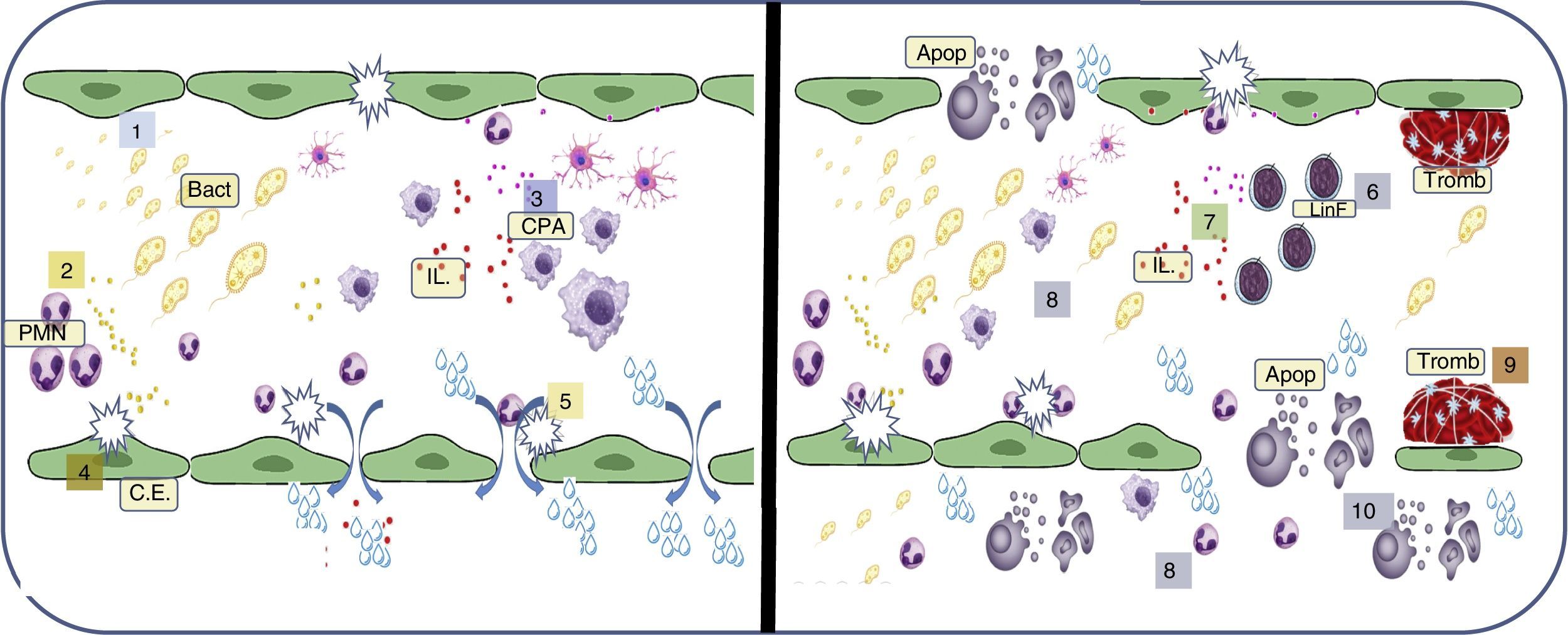
Disfunción endotelial en la sepsisFisiopatológicamente, la sepsis presenta la siguientes etapas: a) infección bacteriana; b) detección de las bacterias por las células residentes; c) inicio de la respuesta inflamatoria; d) activación endotelial; e) aumento de la permeabilidad endotelial; f) alteración de los mecánicos pro/anti-coagulación; g) aumento de los exudados; h) mecanismos vasculares compensatorios; i) fallo de los mecanismos compensatorios; j) hipoxia tisular; k) muerte celular; y k) fallo orgánica múltiple (fig. 2).
 Comienzo de la progresión bacteriana (Bact) en el flujo sanguíneo; 2) respuesta primaria de los polimorfonucleares (PMN); 3) repuesta tardía de células presentadoras de antígeno (CPA); 4) activación de la célula endotelial (CE); 5) aumento de la permeabilidad endotelial; 6) inicio de la respuesta linfocitaria (LinF); 7) desbalance de interleucinas (IL) pro y antiinflamatorias; 8) aumento del estímulo hipóxico endoluminal y exoluminal; 9) formación de trombos secundario al fallo de los mecanismos de anticoagulación; 10) comienzo de la apoptosis (apop) celular.")
Resumen de los procesos vasculares asociados a sepsis. 1) Comienzo de la progresión bacteriana (Bact) en el flujo sanguíneo; 2) respuesta primaria de los polimorfonucleares (PMN); 3) repuesta tardía de células presentadoras de antígeno (CPA); 4) activación de la célula endotelial (CE); 5) aumento de la permeabilidad endotelial; 6) inicio de la respuesta linfocitaria (LinF); 7) desbalance de interleucinas (IL) pro y antiinflamatorias; 8) aumento del estímulo hipóxico endoluminal y exoluminal; 9) formación de trombos secundario al fallo de los mecanismos de anticoagulación; 10) comienzo de la apoptosis (apop) celular.
En la sepsis podemos detectar 2 formas de disfunción vascular. La primera se manifiesta con un compromiso sistémico, donde se caracteriza por una profusa vasodilatación que involucra cambios macrohemodinámicos o de los grandes vasos. Uno de los más evidentes es la reducción en la presión arterial. Y la segunda una alteración en la microcirculación (i.e., arteriolas, capilares y vénulas), en donde la regulación fina de la circulación asociada a la demanda del tejido se ve también comprometida. Si bien estos 2 niveles de disfunción vascular están relacionados, se conoce que alteraciones en el flujo sanguíneo en la microcirculación están asociados a peor pronóstico, al menos parcialmente, de forma independiente de los cambios macrohemodinámicos10. Sin perjuicio de lo anterior ha sido ampliamente mostrado que las alteraciones macro y microvasculares evidenciadas en la sepsis se caracterizan por disfunción de las células endoteliales.
Fisiológicamente las células endoteliales están expuestas a moléculas de señalización circulantes, tales como interleucinas, NO y prostaglandinas, entre otras; y a cambios hemodinámicos o de tensión. En el caso del shock séptico estos mecanismos fisiológicos se ven sobrepasados. Específicamente, el deterioro de la función endotelial en la sepsis conduce a cambios fenotípicos y físicos del endotelio, con liberación desregulada de vasodilatadores potentes, como NO y prostaciclina; reducción de la reactividad vascular a vasoconstrictores, asociada a agregación de leucocitos y plaquetas; aumento de la expresión de iNOS y liberación de nucleósidos —como ATP o ADO— al medio extracelular, entre otros múltiples efectos.
Producto de las alteraciones descritas, la capacidad endotelial de regulación del flujo sanguíneo se reduce y aumenta la permeabilidad de la microcirculación, lo que se traduce en un deterioro de la irrigación tisular. A su vez, estos eventos generan un estado de hipoxia tisular, es decir, una reducción del aporte de oxígeno y nutrientes hacia los tejidos, afectando la viabilidad de estos. Así por ejemplo, en un modelo de ratones sépticos sometidos a ligadura cecal y punción, en los que además se investigó la velocidad del flujo sanguíneo en la extremidad inferior y el contenido de oxígeno en los glóbulos rojos a nivel capilar, se observó un retraso de 3 a 4 veces en el tiempo de respuesta capilar (vasodilatación) dentro de capilares hipóxicos (con glóbulos rojos que contuvieron PO2<20%)11. En el contexto de la sepsis estos resultados indican que el deterioro de la autorregulación vasomotora deriva en reducción en el transporte de O2 hacia el tejido, lo cual puede explicar al menos en parte el fallo múltiple de órganos que ocurre en esta condición patológica.
En este manuscrito queremos hacer énfasis en cómo el cóctel de interleucinas presente en la sepsis puede generar disfunción endotelial. Los trabajos iniciales mostraron que el incremento de interleucinas proinflamatorias disminuían la capacidad de síntesis de NO a partir de la eNOS. Posteriores trabajos mostraron que las interleucinas pro-inflamatorias inducen la expresión de la iNOS, la cual genera cantidades 1.000 veces mayores de NO que la isoforma endotelial, y por tiempos prolongados. De hecho, en la literatura de los años 90 se mencionaba que el exceso de NO generado vía iNOS era el responsable de las complicaciones macrohemodinámicas y microvasculares, características de la sepsis. Estudios más recientes muestran que el TNF-α tiene un efecto disruptivo en la unión de las células endoteliales y además promueve la adhesión de monocitos al endotelio12.
Por otro lado, se ha planteado que en ambientes de estrés la liberación de IL pro-inflamatorias como la IL-6 al medio extracelular traería consigo un aumento del VEGF13. Estos últimos hallazgos son relevantes para el análisis sobre la disfunción microvascular presente en sepsis, ya que VEGF es un factor de crecimiento con múltiples acciones a nivel vascular, que incluyen regulación de la supervivencia endotelial, de la proliferación y migración de células endoteliales y de la formación de vasos sanguíneos; es interesante para el cuadro séptico el incremento de la permeabilidad14. Cabe mencionar que existe una regulación recíproca entre VEFG y NO. Así, VEGF, junto con la iNOS, incrementaría la producción de NO favoreciendo la vasodilatación, el aumento de la permeabilidad en la microcirculación y las complicaciones hemodinámicas asociadas a la sepsis.
Por otro lado, a nivel endotelial se ha reportado que IL-10 tendría un efecto «protector» a los efectos de TNF-α, ya que disminuye la interacción del leucocito-endotelio15. Pero otros resultados asocian la elevación de TNF-α con incremento de P-selectina (proteína que indica lesión en la célula endotelial)16. Por su parte, la IL antiinflamatoria IL-4 aumenta la permeabilidad vascular en arterias coronarias17. En consecuencia las interleucinas tanto pro como antiinflamatorias modulan la función del endotelio durante la sepsis.
Hasta ahora hemos hecho énfasis en cómo en un cuadro de sepsis incrementaría la disfunción vascular traducida por un exceso en la síntesis de NO, incremento de la permeabilidad vía VEGF, lo que lleva a edema, compromiso de la perfusión tisular y generación de un círculo vicioso potenciador. Ya se ha mencionado que en sepsis existe un estado de hipoxia tisular. Bajo esta condición se incrementa el nivel extracelular del nucleósido adenosina (ADO). Dado que el foco de la revisión es resaltar la vía A2A-NO-VEGF en la sepsis, nos enfocarnos en la participación de A2A.
Adenosina, receptor A2A e inmunosupresiónLa adenosina vía activación de sus receptores de membrana acoplados a proteína G (A1, A2A, A2B y A3) tiene efectos pleiotrópicos en el sistema cardiovascular e inmunológico. Por ejemplo, en el sistema inmune se ha documentado que ADO altera la diferenciación de células dendríticas afectando la efectividad de los linfocitos CD8+, induciendo un estado tolerogénico18. Más específicamente, los receptores A2A de adenosina están presentes en células presentadoras de antígeno, linfocitos T reguladores (CD4+, CD25+), linfocitos T ejecutores (CD8+) y linfocitos B en las cuales pueden aumentar la producción de interleucinas antiinflamatorias como también intrínsecamente disminuir la capacidad efectora de estas células19. En este contexto, Bao et al.20 demostraron cómo ADO, vía activación de A2A, estimula a linfocitos T reguladores (Treg) durante la respuesta séptica en ratones. Además, Li et al.21 en un modelo de una enfermedad autoinmune, como la miastenia gravis, encontraron que la estimulación del A2A en linfocitos B, presentes en el bazo y en los ganglios linfáticos, inhibe la producción in vitro de anticuerpos anti-acetilcolina. Con ello podemos indicar que A2A tiene un rol preponderante en la regulación del sistema inmunológico.
Revisaremos más en detalle el rol de A2A sobre los Treg y cómo genera inmunosupresión. En este sentido Hank et al.22 observaron que al tratar ratones con los agonistas del receptor A2A, ATL146e, ATL370 y ATL1223 y que fueron trasplantados con células madre hematopoyéticas obtenidas de donantes, se logró suprimir el rechazo del injerto y el cuadro inflamatorio en el huésped. El mecanismo detrás de este fenómeno sería el aumento, tanto en sangre como en tejido, de los linfocitos Treg derivados de las células donantes, que ocurre secundario a la activación de A2A22. A su vez, otro estudio23 utilizando un modelo de ratas con trasplante hepático, tratadas o no con un agonista del receptor A2A (CGS-21680) o un antagonista del mismo (ZM-241385), encontraron que CGS-21680 disminuyó la expresión de las proteínas asociadas a disfunción vascular y proinflamación, tales como TNF-α, proteína inflamatoria macrófagos 2 (MIP-2) y la molécula de adhesión intercelular 1 (ICAM-1).
Complementando lo anterior, la inmunosupresión ejercida por A2A fue también confirmada aumentando el número24 y la función de células CD4+25. Además se ha demostrado que estas células poseen la capacidad intrínseca de metabolizar el ATP en ADO generando un estado sinérgico inmunosupresivo. La excesiva estimulación de A2A, esperada en sepsis, podría llevar a un estado de «inmunoplejía», caracterizada por una menor capacidad de respuesta del sistema inmune ante la presencia de bacterias25. La relación entre ADO, A2A y la producción de citoquinas pro y antiinflamatorias en el marco de la sepsis es conocida parcialmente. Así, A2A controla la expresión de IL-10 en macrófagos en ambientes sépticos, como mostraron Csoka et al.26 usando un modelo de ratón infectado por Escherichia coli (E. coli). Estos autores reportan que la activación del A2A participa en el control de la expresión de IL-10 a nivel de la activación del gen que codifica para esta proteína (i.e. regulación transcripcional).
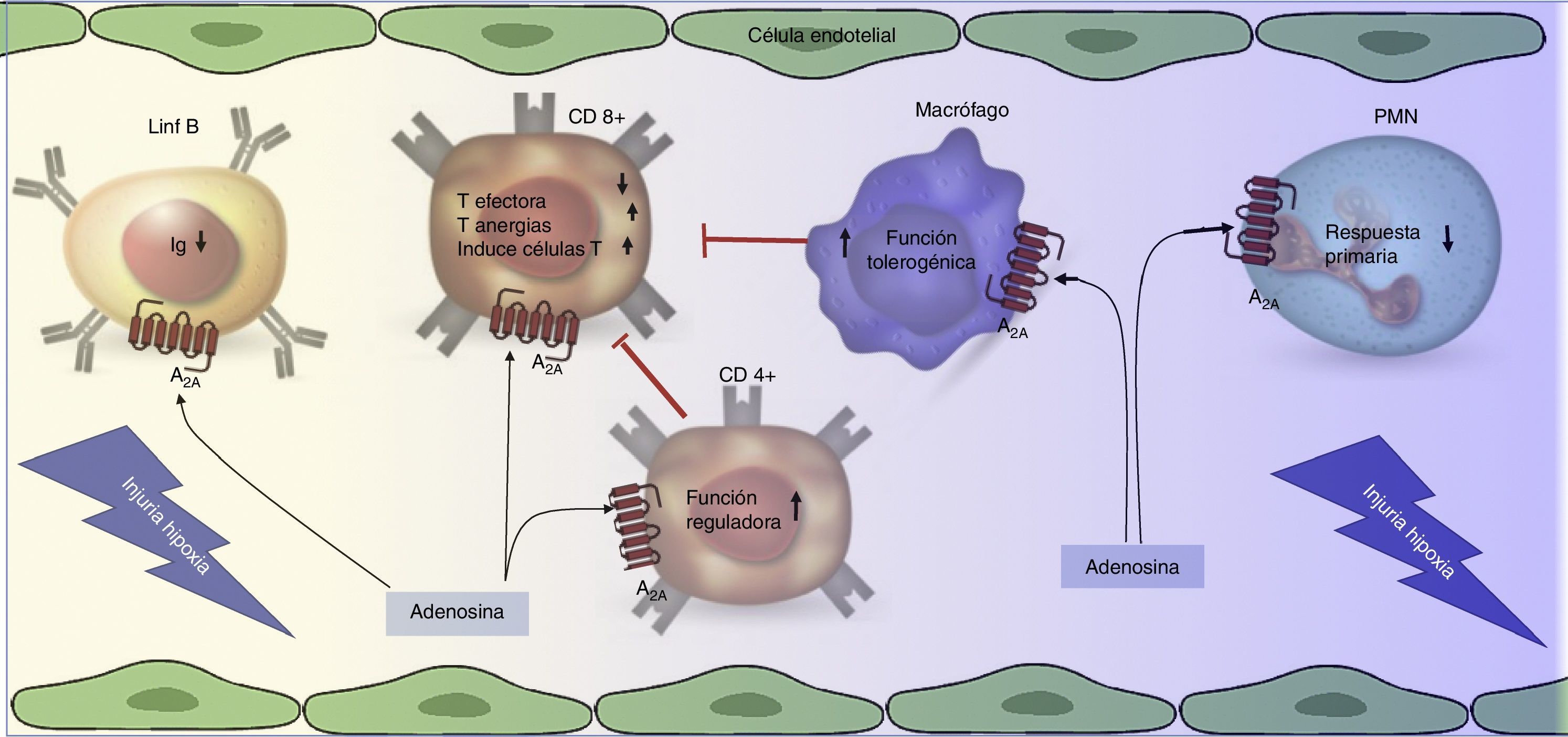
La evidencia más significativa respecto a la importancia del receptor A2A en sepsis proviene de estudios in vivo utilizando ratones deficientes en A2A (A2AKO). Así, en un estudio publicado por Alam et al.27 en el que se utilizaron ratones A2AKO y control (wild type [WT]), se pudo observar que al administrar el agonista ATL313 en ratones control se redujo el cuadro inflamatorio y la gastritis, pero aumentó la carga bacteriana de Helicobacter pylori. Por el contrario, en los ratones A2AKO aumentó la gastritis pero se limitó la infección. Además, otros resultados de este mismo estudio indican que la activación del receptor A2A limita la liberación de moléculas proinflamatorias, inhibe la respuesta efectora y aumenta la respuesta reguladora del sistema inmune. En este mismo contexto Nemeth et al.28 desarrollaron un modelo de ratón séptico mediante el procedimiento de ligación cecal y punción en ratones A2AKO, vale decir un modelo de infección multibacteriana. La supervivencia del A2AKO en este modelo fue cercana al 70% a los 5 días, mientras que fue de solo del 30% en los ratones WT. Este efecto se asocia a un menor recuento de unidades formadoras de colonias en sangre y lavado peritoneal a las 16horas en el ratón A2AKO. Por otro lado, se evaluó las diferencias en las concentraciones de interleucinas séricas de ambos grupos. Se observó que el ratón WT presentó una elevada concentración de IL-10 sérica respecto al ratón A2AKO. A partir de estos resultados se concluye que A2A genera una inhibición en la respuesta inmune celular y humoral frente a patógenos, lo cual disminuye el aclaramiento de la carga bacteriana, tanto en sangre como en tejidos. El resumen de estos hallazgos y cómo participaría A2A en la sepsis se resume en la figura 3.
 durante el control de la función de células inmunológicas. El daño hipóxico genera un aumento de la producción de adenosina. Esta molécula al hacer blanco en el receptor A2A (A2A), presente en células del sistema inmune, reduce su función. Al mismo tiempo al activarse el A2A en los linfocitos T reguladores (CD4+) se estimula su función reguladora, lo que disminuye la respuesta de los linfocitos T ejecutores (CD8+).")
Resumen de la función del receptor A2A (A2A) durante el control de la función de células inmunológicas. El daño hipóxico genera un aumento de la producción de adenosina. Esta molécula al hacer blanco en el receptor A2A (A2A), presente en células del sistema inmune, reduce su función. Al mismo tiempo al activarse el A2A en los linfocitos T reguladores (CD4+) se estimula su función reguladora, lo que disminuye la respuesta de los linfocitos T ejecutores (CD8+).
En otro modelo Sullivan et al.29 muestran que la sobrevivencia de ratones inyectados con LPS aumentó en forma dosis dependiente al administrar el agonista A2A, ATL 146e. Para confirmar este hallazgo estos autores utilizaron un antagonista selectivo del A2A o ratones A2AKO. En ambos grupos se encontró un incremento en la mortalidad de los animales tratados con LPS. Adicionalmente, en el mismo trabajo, el grupo usó un modelo in vivo de daño con E. coli intraabdominal donde compararon la supervivencia de 4 grupos de ratones: 1) control de infectados con E. coli; 2) infectados con E. coli+antibiótico ceftriaxona; 3) infectados E. coli+ATL 146e; y 4) infectados E. coli+ATL 146e+ceftriaxona. Los resultados mostraron que el grupo infectado con E. coli que recibió ATL 146e+ceftriaxona tuvo mejor supervivencia en comparación con los otros grupos a las 72horas postinoculación. Esta variación en la supervivencia estuvo asociada con un menor recuento de unidades formadoras de colonias en sangre. En consecuencia, acorde a estos resultados A2A facilitaría la función inmunológica ante E. coli o LPS.
Lo hallado por Sullivan et al.29 es refrendado por lo realizado por Moore et al.30, quienes además de usar LPS y E. coli usaron Staphylococcus aureus en un modelo de ratón séptico con inoculación intraabdominal. Ellos mostraron que al usar el agonista del A2A, ATL313, en conjunto con antibiótico, la supervivencia era del 100%, mientras que únicamente fue del 20% en el grupo donde se usó solo antibiótico. Esta diferencia está asociada a reducción en el nivel de interleucinas proinflamatorias TNF-α, MIP-1 e IFN-γ en el grupo tratado con ATL313.
En conjunto estos resultados plantean el efecto sinérgico del antibiótico y los agonistas A2A que disminuiría la carga bacteriana y mejora la supervivencia, al menos en modelos animales de sepsis. Esto también se ha mostrado en modelos endotoxémicos que usan LPS. Sin embargo, los modelos endotoxémicos no logran explicar a cabalidad el desarrollo de sepsis, dado que no consideran la presencia de bacterias, una variable que como hemos visto es fundamental para comprender el comportamiento del sistema inmune en el contexto de la sepsis.
A pesar de la relevancia de estos estudios resaltamos también el hecho de que ellos no exploran la presencia de daño de células endoteliales en los distintos modelos de sepsis. Pensamos que es una limitación, ya que no logra visualizar la importancia que ADO, y en particular el receptor A2A, tendría sobre este sistema. Por lo mismo, en la siguiente sección haremos énfasis en la literatura disponible sobre la participación de estas moléculas en la función vascular durante la sepsis.
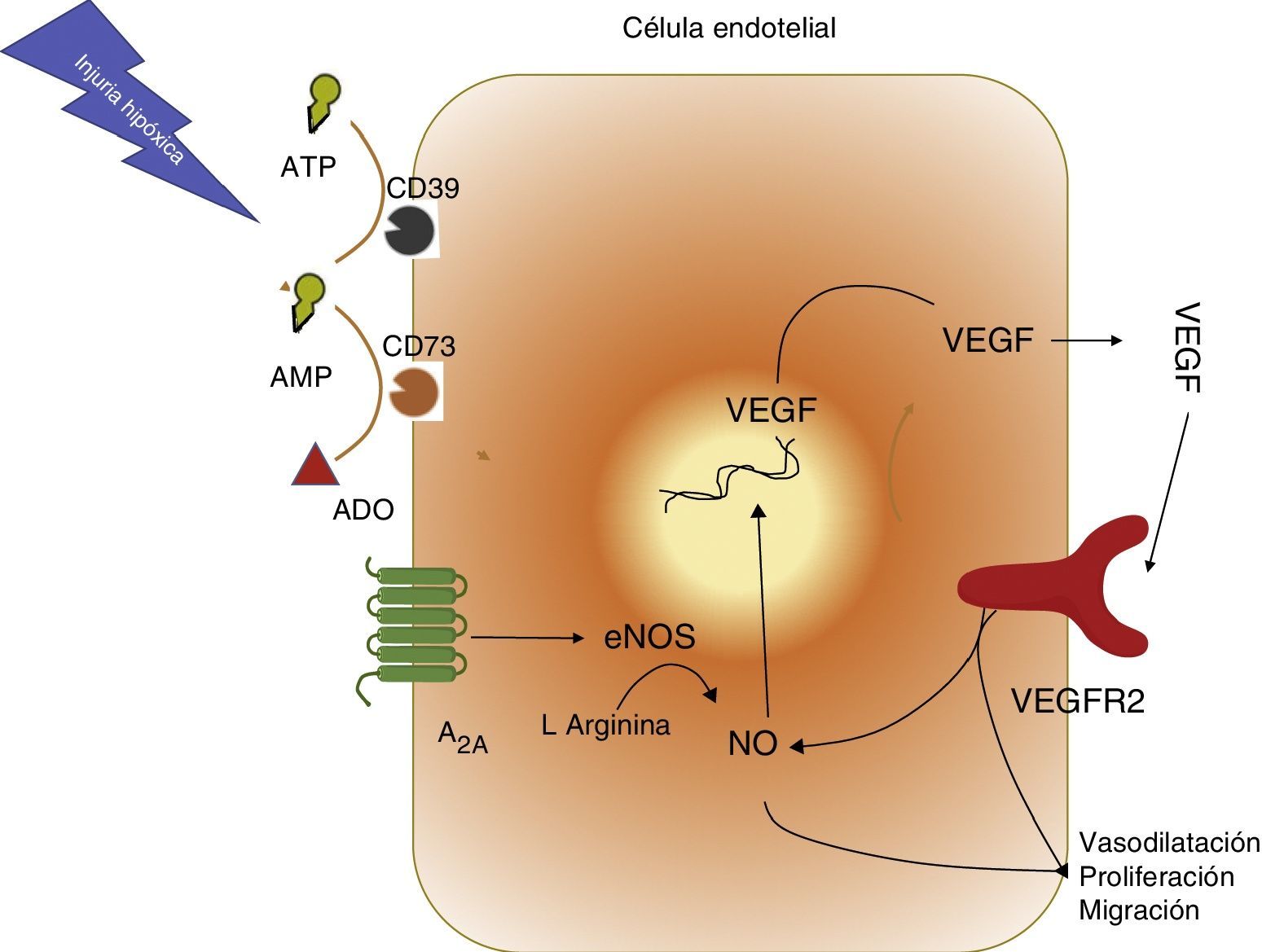
A2A-óxido nítrico-factor de crecimiento de endotelio vascular en la sepsisVarias investigaciones31,32, incluyendo algunas de nuestro grupo7,8, han mostrado que la activación del receptor A2A llevaría a un aumento en la síntesis de NO y de VEGF (fig. 4). El objetivo de esto sería establecer nuevas redes de microcirculación con el fin de mantener la oxigenación en los tejidos hipóxicos, como sucede por ejemplo en la sepsis. Sin embargo, pese a que inicialmente esta vía sería reconocida como «protectora» o «fisiológica» si permanece activa, como por ejemplo podría ocurrir en la sepsis, sería también la responsable del daño vascular multisistémico.
 y factor de crecimiento de endotelio vascular (VEGF). Se resumen los hallazgos respecto a la interacción entre adenosina (ADO), el receptor A2A (A2A), NO sintasa endotelial (eNOS), VEGF y su receptor de tipo 2 (VEGFR2). Se resalta el eje central de NO para generar vasodilatación y formación de nuevos vasos sanguíneos.")
Vía de activación receptor A2A, óxido nítrico (NO) y factor de crecimiento de endotelio vascular (VEGF). Se resumen los hallazgos respecto a la interacción entre adenosina (ADO), el receptor A2A (A2A), NO sintasa endotelial (eNOS), VEGF y su receptor de tipo 2 (VEGFR2). Se resalta el eje central de NO para generar vasodilatación y formación de nuevos vasos sanguíneos.
La información sobre ADO, A2A, NO y VEGF en sepsis es amplia si se busca cualquiera de estas moléculas de forma individual. Sin embargo, no logramos encontrar evidencia en búsquedas que incluyan todas ellas. Únicamente, a modo de ejemplo indicaremos que la liberación exagerada de NO vía iNOS en cuadros sépticos generaría una vasodilatación exagerada a nivel renal alterando su irrigación, lo que aumentaría el daño tubular renal característico en cuadros sépticos. En el contexto de la sepsis Tsukahara et al.9 mostraron que los neutrófilos de pacientes sépticos expresaron niveles más altos de iNOS respecto al grupo de neutrófilos de pacientes sanos luego de la estimulación con LPS. En apoyo a esto se sabe que el sustrato para la síntesis de NO es el aminoácido semiesencial L-arginina, el cual está disminuido en individuos con sepsis respecto a los controles sanos33, sugiriendo un alto «consumo» del aminoácido para síntesis de NO. Adicionalmente, se ha demostrado que LPS e interleucinas, tales como TNF-α, IL-1 e interferón gama (INF-ϒ)34 inducen la iNOS en el endotelio, así como en células del músculo liso vascular, los macrófagos y las diferentes células parenquimatosas. En consecuencia la síntesis de NO vía iNOS está incrementada en sepsis, lo cual da cuenta del estado de vasodilatación y explica al menos en parte la fisiopatología del shock séptico.
Por otro lado, la estimulación de A2A por CGS-21680 inhibe la expresión de iNOS y la síntesis de NO inducida por INF ϒ, TNF-α e IL-1β en células HUVEC y células C635, sugiriendo que A2A no solo regularía la actividad de eNOS, en estados «fisiológicos», sino también podría regular la expresión de iNOS en estados patológicos. Los mecanismos celulares de esta última asociación no se conocen. Pero se ha descrito que el incremento de la expresión de iNOS mediado por interleucinas proinflamatorias fue inhibido por un activador de la adenil-ciclasa (AC) (recordar que los receptores A2A también activan AC) o por inhibición de la adenosina quinasa (que degrada ADO en el extracelular) (ver detalles en Escudero y Sobrevia36). Estos resultados complementan datos previos que muestran que la sobreexpresión de A2A reduce la expresión de iNOS estimulada por LPS, INF-ϒ y TNF-α en células C635. No se conoce cómo sería la interregulación de A2A-eNOS o A2A-iNOS en el caso de la sepsis, pero es probable que desde una regulación fisiológica con una síntesis controlada de NO, mediado por la interacción A2A-eNOS, se llegue a un estado compensatorio A2A-dependiente en el que se busque contrabalancear la sobreexpresión de iNOS estimulada por un microambiente inflamatorio. Esta hipótesis debe ser probada.
Por otro lado, Leibovich et al.37, usando macrófagos de ratones estimulándolos con NECA (agonista no selectivo de los receptores de adenosina) o con CGS-21680, encontraron que se generaba un aumento significativo de la expresión de VEGF. En estas mismas condiciones, al tratar los macrófagos con LPS o INF-ϒ se determinó que no se generó cambio en la producción de VEGF. Los autores muestran que al utilizar LPS+NECA o CGS21680 se encontró un efecto sinérgico potenciador, ya que existía un aumento de 10 veces en la producción de VEGF. Se determinó además mediante un ensayo de angiogénesis que este VEGF es biológicamente activo37. De igual forma a lo que sucede con iNOS se desconoce cómo el eje regulatorio A2A-NO-VEGF se modificaría en el estado de sepsis. En esta condición de sepsis existiría una mayor expresión de iNOS y VEGF, que por un lado incrementaría la síntesis de NO, pero también podría existir un mecanismos contrarregulatorio. Además, no se conoce cómo se encajaría en este complejo escenario regulatorio el rol de ADO y particularmente del A2A en la funcionalidad vascular. Es de esperarse que, a nivel vascular, el A2A tienda a reponer un estado fisiológico.
De igual importancia Nemeth et al.28 mostraron que la estimulación del receptor A2A en un modelo de sepsis polimicrobiana abdominal generaría elevados niveles de IL-10, siendo esto perjudicial para los ratones sépticos, que mostraron altos niveles de mortalidad. Esto podría relacionarse con datos que muestran que esta interleucina inhibiría la expresión de NO38, lo que torna aún más compleja la comprensión del rol de ADO y el receptor A2A en la regulación de la producción de NO y VEGF, como también en la regulación de la función de iNOS. Por ende, es relevante que se hagan los esfuerzos necesarios para la comprensión de dichos elementos generando una nueva alternativa de manejo en esta compleja afección.
Conclusión y direcciones futurasPodríamos concluir que la adenosina vía activación de sus receptores A2A tiene efectos pleiotrópicos en el sistema cardiovascular e inmunológico. De lo revisado se desprende que el receptor A2A genera un estado de inmunosupresión asociado a una reducción en la síntesis de interleucinas proinflamatorias, así como en la actividad de linfocitos CD8+, macrófagos y polimorfonucleares. La evidencia en modelos animales A2AKO es contradictoria respecto a la mortalidad de estos animales expuestos a sepsis. Las causas se asocian al modelo de sepsis utilizado, siendo unibacteriano, multibacteriano o con inyección de LPS. Además, en esta revisión también se concluye que el NO desempeña un rol significativo en el mantenimiento de la integridad órgano/vascular, ligado a la respuesta vasodilatadora. En la sepsis se generaría un exceso de NO vía iNOS, lo que sería fundamental en la vasodilatación exacerbada evidenciada en cuadros sépticos. En cambio, podríamos pensar que en estados hipóxicos la liberación de ADO y la activación del receptor A2A provocarían una liberación de NO que podría ejercer un efecto protector frente a dichos estados hipóxicos aumentando el flujo vascular en órganos afectados.
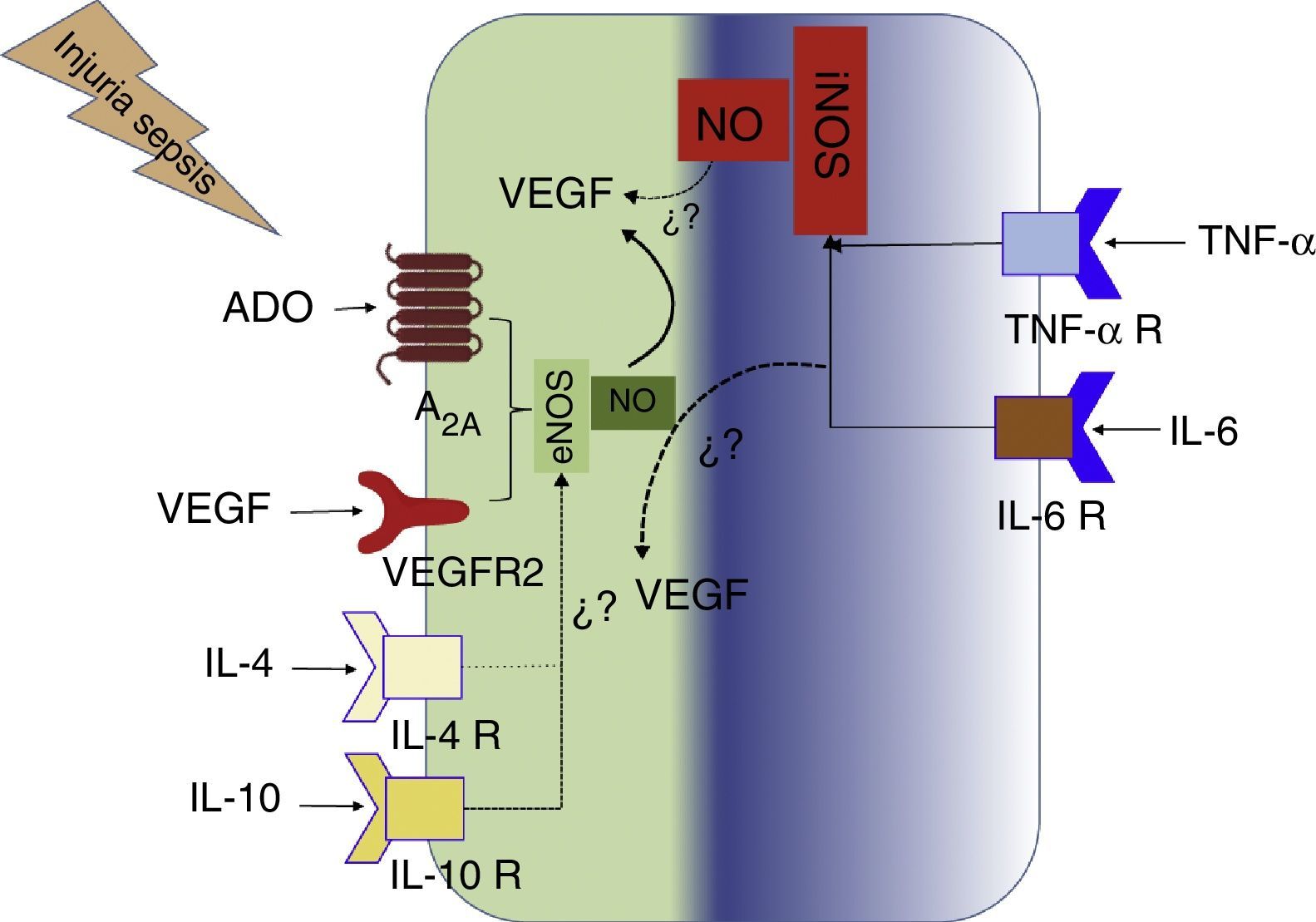
También hay que considerar que A2A, eNOS, iNOS y VEGF son proteínas que incrementan su expresión en hipoxia, o por interleucinas proinflamatorias. Por otro lado se ha reportado que varias moléculas presentes en la sepsis inducirían la activación de eNOS, y por ende la producción de NO, tal es el caso de IL-6, TNF-α y LPS39. Paradójicamente se ha reportado que IL-10 también activaría la función de eNOS y la producción de NO40. Por consiguiente, en este ambiente observado en pacientes sépticos todas estas proteínas se encontrarían sobreexpresadas. Cómo participan estas proteínas en la generación de la disfunción vascular evidenciada en sepsis no es conocido. En esta revisión proponemos un modelo de trabajo en la cual sugerimos la interacción de las A2A, eNOS, iNOS, VEGF y citoquinas en el cuadro séptico (fig. 5). Así, en el medio séptico/hipóxico la célula endotelial se ve sometida a diversos estímulos, entre ellos la estimulación de A2A secundaria a la liberación masiva de ADO, lo cual activaría la eNOS que cataliza la producción de NO en cantidades nanomolares. Fisiológicamente, este gas genera vasodilatación e incrementa la expresión del VEGF. Este VEGF a su vez tendría un efecto de retroalimentación positiva en la producción de NO vía activación de VEGFR2. Por otro lado, en una condición de sepsis las moléculas proinflamatorias tales como TNF-α e IL-6 activan la expresión de la iNOS, la cual produce NO en el rango micromolar. Este efecto perpetúa la vasodilatación y las alteraciones distributivas en estados sépticos. A su vez, en sepsis también incrementa la expresión de citoquinas antiinflamatorias como IL-10 e IL-4. Si bien se conoce que A2A inhibe la expresión de iNOS y que TNF-α e IL-6 provocan disfunción endotelial asociada a desregulación de la eNOS, se desconoce la interacción entre TNF-α e IL-6 con VEGF, o entre IL-10 e IL-4 con eNOS o VEGF. Este modelo es hipotético, por lo que es necesario realizar estudios encaminados a dilucidar lo propuesto.
Financiación- factor de crecimiento de endotelio vascular (VEGF) en sepsis.ADO: adenosina; eNOS: óxido nítrico sintasa endotelial; IL: interleucina;TNF-α: factor de necrosis tumoral α.")
Este estudio fue financiado por Fondecyt Regular 1140586; Fondequip EQM140104; DIUBB 166709 3/R; y GI 171709/VC.
Conflicto de interesesNinguno.
Nos gustaría agradecer a los investigadores del Laboratorio de Fisiología Vascular, al Grupo de Investigación en Angiogénesis Tumoral (GIANT) y al Grupo de Investigación e Innovación en Salud Vascular (GRIVAS Health) por las interesantes discusiones e ideas respecto a este manuscrito.
