


El síndrome de Ehlers-Danlos de tipo IV, vascular o de Sack-Barabas (SED-IV), es una conectivopatía muy rara, de transmisión autosómica dominante, originada por una mutación del gen COL3A1, que provoca un déficit de colágeno de tipo iii1. Esto ocasiona una fragilidad tisular allí donde está presente: piel, tubo digestivo, paredes vasculares, etc.2.
Presentamos el caso dramático, de una paciente, en el que mostramos la rápida aparición de las complicaciones del síndrome, que le ocasionó la muerte, aunque sospechamos el diagnóstico desde el principio.
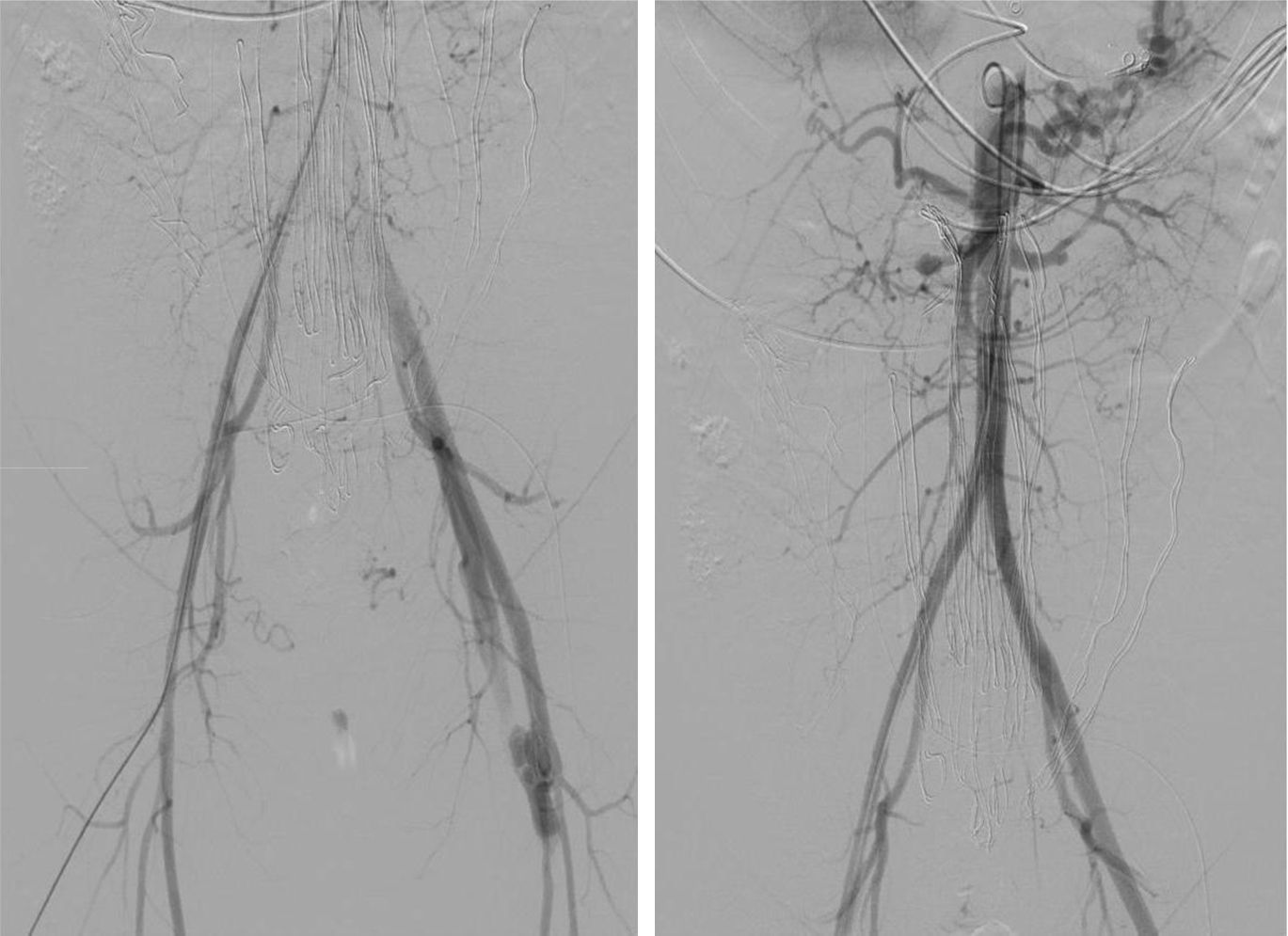
Mujer de 27 años con antecedentes de hepatitis autoinmune y déficit del factor VIII y XIII de la coagulación, que ingresó en urología tras acudir en 2 ocasiones a urgencias con clínica de dolor en fosas renales, afebril, hematuria, leucocitosis, sin anemización y estudio ecográfico inespecífico. Al tercer día de la hospitalización sufrió hipotensión y abdomen agudo, con Hb de 7,2g/dl y en la TAC urgente se descubre hemoperitoneo. Se intervino por cirugía general, que encontró hematoma en el meso, con sufrimiento de un segmento de intestino delgado, que obligó a su resección. La paciente pasó a la UCI e inmediatamente, ante la duda de no haber localizado el sangrado, se realizó arteriografía urgente que mostró: ausencia de sangrado activo, existencia de fístula arteriovenosa femoral izquierda, tortuosidad y dilatación de arterias renales (fig. 1). Al regresar la paciente a la UCI sufrió un cuadro de shock hipovolémico (Hb: 5,0g/dl), por lo que nuevamente fue intervenida de forma inmediata, encontrándose sangrado activo por la arteria ilíaca izquierda, que obligó a su ligadura para controlar dicha hemorragia.

Es tras este gesto quirúrgico cuando es avisado el equipo de cirugía vascular, que a la llegada al quirófano constata que el sangrado está controlado, pero la pierna izquierda está en grave isquemia. Se revisan los hallazgos de la TAC y la arteriografía, y se observa el fenotipo de la paciente (piel del tórax traslúcida, dejando ver llamativamente la red venosa subyacente, nariz puntiaguda, orejas hipolobuladas). Todo ello hace sospechar que nos encontramos ante una conectivopatía con afectación vascular grave.
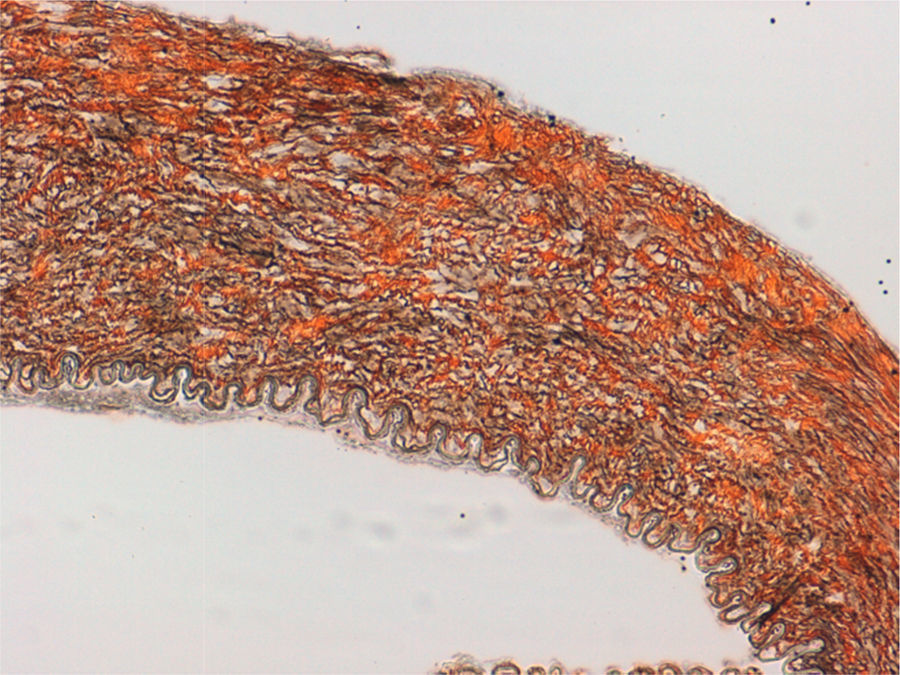
Dada la necesidad de revascularizar la pierna izquierda, se decide realizar un bypass fémoro-femoral, previa explicación de la gravedad de la situación y las posibles complicaciones a sus padres. El acto quirúrgico fue muy complejo porque el simple gesto de control de las arterias, las rompía, y el sangrado fue constante durante la realización de las distintas anastomosis: previo al bypass fémoro-femoral, fue preciso realizar un bypass desde la arteria ilíaca externa hasta bifurcación femoral derecha para solucionar la rotura de la arteria femoral ocasionada durante el clampaje. Se tomó muestra de segmentos arteriales para anatomía patológica.
La paciente regresó a la UCI estable, con las piernas bien perfundidas. Se solicitó estudio genético de la paciente ante la alta sospecha de SED-IV. El resultado de la anatomía patológica de los segmentos arteriales fue compatible con SED-IV (fig. 2).

La paciente evolucionó inicialmente bien, y permaneció estable hasta el 8.° día, en el que sufrió parada cardiaca con signos de IAM y hemopericardio. Pese a las maniobras de resucitación, la paciente falleció.
Se realizó autopsia clínica con el siguiente resultado: a) Infarto de miocardio masivo en ventrículo izquierdo con taponamiento cardiaco por hemopericardio intenso y rotura de arteria descendente anterior como causa inmediata de la muerte; b) Hemotórax bilateral; c) Hemoperitoneo residual; d) Hematoma disecante en ambas arterias renales; e) Vejiga con trama vascular marcada y hemorragias. Todo ello compatible con SED-IV como enfermedad fundamental.
Finalmente se recibió el informe del estudio genético solicitado que confirmó que la paciente era portadora de la variante patogénica c.3266G>A en el GEN COL3A1. Variante patogénica asociada al SED-IV. Se solicitó estudio familiar.
El síndrome de Ehlers-Danlos incluye un grupo heterogéneo de enfermedades del tejido conectivo3. Una de ellas es el tipo IV, vascular o síndrome de Sack-Barabas. Entidad muy rara, y con una expectativa de vida corta debido a las complicaciones que provoca. La sospecha diagnóstica es fácil si se sabe de su existencia: aparición de rotura arterial, intestinal o uterina en paciente joven (menor de 40 años), con el fenotipo característico y que curiosamente carece de la hiperelasticidad cutánea y articular que tienen el resto de tipos del síndrome (esto puede confundir el diagnóstico a quien nunca se ha enfrentado a este síndrome). Cuando se sospeche se solicitará estudio molecular-genético.
La experiencia internacional se basa en casos y series pequeñas. Una revisión4 identificó 231 pacientes. El 40% presentaban aneurismas. En el 33% hubo rotura arterial sin aneurisma subyacente. La mortalidad poscirugía fue del 30% frente al 24% postratamiento endovascular, por lo que si es posible, se debe optar por esta modalidad terapéutica. En nuestro caso, no pudimos porque intervenimos cuando ya habían ligado la arteria ilíaca, aunque esto no influyó en la muerte de la paciente. La edad media de los pacientes en el momento de su fallecimiento fue de 31 años. Solo en el 24% de los casos recientes se verificó el defecto genético.
En España, los casos publicados son muy escasos5–7. El artículo publicado por Escribano et al. tiene muchas semejanzas al nuestro7.
En conclusión, el SED-IV es un trastorno grave con elevada mortalidad, sin tratamiento curativo, donde la cirugía solo debe indicarse en situaciones de riesgo vital y en el que la terapia endovascular no ha mejorado su pronóstico.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que en este trabajo no se han realizado experimentos en personas y animales.
Confidencialidad de los datosLos autores declaran que en este artículo se han seguido los protocolos del centro sobre la confidencialidad.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no se muestran datos de la paciente.
