




Objectives. Filibuvir is a non-nucleoside inhibitor of hepatitis C virus (HCV) polymerase. This study evaluated the safety and efficacy of filibuvir plus pegylated interferon alfa-2a (pegIFN)/ribavirin.
Material and methods. Treatment-naïve, HCV genotype-1 patients were randomized to receive filibuvir 300 or 600 mg twice daily (BID) or placebo plus pegIFN (180 Mg/wk) and ribavirin (1,000/1,200 mg BID) for 24 weeks. Filibuvir patients who achieved defined response through week 24 discontinued therapy at week 24. All other patients continued on open-label peglFN/ribavirin through week 48. The primary endpoint was the proportion of patients who achieved sustained virologic response (SVR) defined as HCV RNA < 15 IU/mL at end of treatment (weeks 24 or 48) and week 72.
Results. Overall, 288 patients were randomized and treated. SVR was achieved by 41.7, 39.6, and 45.8% of patients in the filibuvir 300 mg, 600 mg, and placebo arms, respectively. While the addition of filibuvir to pegIFN/ribavirin improved on-treatment virologic response parameters, this did not translate into improved SVR rates due to a high rate of virologic relapse following completion of therapy (300 mg: 35.9%; 600 mg: 42.9%; placebo: 25.4%). The most commonly reported adverse events were nausea, fatigue, headache, and insomnia, and were reported at similar rates across arms. Conclusions. Filibuvir plus pegIFN/ribavirin did not improve the percentage of patients achieving SVR compared with administration of pegIFN/ribavirin alone. However, the agent was well tolerated and was associated with higher on-treatment virologic response parameters. Further evaluation of filibuvir in combination with other direct-acting antiviral agents may be considered.
Filibuvir (FLV; Pfizer Inc, New York, NY, USA) is a non-nucleoside inhibitor (NNI) of the hepatitis C virus (HCV) ribonucleic acid (RNA)-dependent RNA polymerase. In vitro, FLV exhibits potent activity against HCV polymerases and replicons derived from genotype 1 strains of HCV, with equipotent activity against the 1a and 1b subtypes.1 Initial studies in HCV-infected patients demonstrated a dose-dependent reduction in HCV RNA concentrations following administration of FLV monotherapy for 7-10 days, with mean maximum reductions of > 2.0 log10 IU/mL at doses > 450 mg twice daily (BID).2 When administered in combination with pegylated interferon alfa-2a (pegIFN) and ribavirin (RBV) for 4 weeks, a significant increase in the rate of rapid virologic response (RVR) was observed compared with administration of pegIFN/RBV alone (60-75% vs. 0%, respectively); however, continued follow-up of patients determined that the initial response to FLV therapy did not translate into higher rates of sustained virologic response (SVR), and it was hypothesized this was due to the short duration of FLV administration.3
The present study was conducted to determine whether longer duration of FLV administered in combination with pegIFN/RBV would significantly increase the proportion of patients who achieved SVR. This was a multicenter, multinational, double-blind, randomized, placebo-controlled Phase 2b study to assess the safety and efficacy of FLV dosed at 300 or 600 mg BID in combination with pegIFN and RBV in treatment-naïve HCV genotype 1 infected patients.
Material and MethodsPatientsAll patients provided written informed consent. The study is registered with ClinicalTrials.gov (NCT00987337). The protocol and informed consent forms were approved by independent ethics committees at all study centers in accordance with national procedures. The studies were conducted according to the ethical principles in the Declaration of Helsinki4 and in compliance with all Good Clinical Practice guidelines, local laws, and regulations.
Patients from 70 sites in North America (United States and Canada), Europe (Belgium, France, Germany, Hungary, and Spain), and Asia (Republic of Korea) were screened for inclusion in the study. Treatment-naïve (no prior treatment with IFN ± RBV or investigational anti-HCV agents) male and female patients aged ≥ 18 years were eligible for inclusion in the study. All patients were required to be HCV seropositive, infected with a genotype 1 strain (VERSANT HCV Genotype 2.0 Assay, Innogenetics, Ghent, Belgium), and have plasma HCV RNA levels ≥ 10,000 IU/mL at screening. In addition, a non-cirrhotic fibrosis classification (i.e., Ishak score ≤ 4 or equivalent) from a liver biopsy obtained within 24 months of screening was required for enrollment.
Patients were excluded if they were co-infected with either HIV or hepatitis B, had evidence of severe or decompensated liver disease or liver disease unrelated to HCV infection, or had any pre-existing medical condition or laboratory abnormality that made them unsuitable for treatment with pegIFN/ RBV. Additional exclusion criteria included an abnormal electrocardiogram (ECG) suggestive of clinically significant cardiac disease or QTc > 450 ms at screening, and history of solid organ transplant, or active alcohol or substance abuse sufficient to prevent adherence to study medication and/or follow-up. Lastly, female patients who were pregnant or nursing and male patients whose female partner was pregnant were excluded.
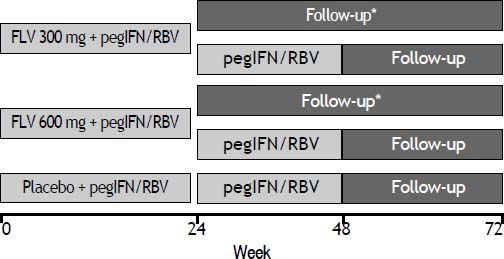
Study designPatients were randomized in a 1:1:1 ratio to receive FLV dosed at 300 or 600 mg BID or matching placebo in combination with pegIFN/RBV, and were stratified according to screening HCV RNA level (≤ 400,000 IU/mL or > 400,000 IU/mL) and by participation in a semen sub-study. FLV or matching placebo was administered during the first 24 weeks of the study only. Those patients randomized to either of the FLV arms who achieved HCV RNA < 15 IU/mL by week 4 and whose HCV RNA level remained < 15 IU/mL through week 24 discontinued all therapy at week 24. All other patients continued on open-label pegIFN/ RBV through week 48 (Figure 1). Patients and investigators were unblinded to treatment assignment at week 24 to determine eligibility to discontinue therapy.

Study schematic. *Patients randomized to either of the FLV groups who achieved HCV RNA < 15 IU/mL by week 4, and whose HCV RNA level remained < 15 IU/mL through week 24 discontinued all therapy at week 24. All other patients continued on oped-label pegIFN/RBV through week 48. FLV: filibuvir. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin.
All patients had a screening visit up to 28 days before the start of dosing; visits at baseline, weeks 2 and 4, and monthly visits thereafter through week 24. Patients who discontinued therapy at week 24 had follow-up visits at weeks 36, 48, and 72. Patients who required therapy beyond week 24 continued to have monthly visits through week 48, and follow-up visits at weeks 60 and 72.
An independent data monitoring committee was responsible for periodic assessments of safety and efficacy data, and made recommendations concerning continuation, termination, or other modifications to the trial based on the observed beneficial or adverse effects of the treatment under study. All sponsor personnel responsible for the conduct of the trial, with the exception of the sponsor study programmer, remained blinded to the results provided to the data monitoring committee.
Stopping rulesPatients who met the following criteria discontinued FLV/placebo but were allowed to remain on pegIFN/RBV at the discretion of the investigator: <1 log reduction in HCV RNA relative to baseline by week 4; HCV RNA > 15 IU/mL at week 12; or > 2 log increase in HCV RNA from nadir. Patients who met any of the following criteria were required to discontinue all therapy (FLV/placebo, pegIFN, and RBV): < 2 log reduction in HCV RNA relative to baseline by week 12; HCV RNA > 15 IU/mL at week 24; two or more consecutive HCV RNA measurements > 1,000 IU/mL following achievement of HCV RNA < 15 IU/mL. Decisions to reduce the dose of pegIFN or RBV, temporarily interrupt, or permanently discontinue therapy in patients experiencing laboratory abnormalities, adverse events (AEs), or serious AEs (SAEs) were made by individual investigators in accordance with the guidelines contained in the prescribing information for pegIFN and/or RBV. Any temporary interruption or permanent discontinuation of pegIFN/RBV required a similar interruption or discontinuation of FLV/placebo to avoid treatment of patients with FLV monotherapy.
EndpointsThe primary endpoint for the study was the proportion of patients who achieved SVR, defined as HCV RNA < 15 IU/mL at both end of treatment (Weeks 24 or 48) and Week 72. Key secondary endpoints included the proportion of patients with HCV RNA < 15 IU/mL at week 4 (RVR), week 12 (complete early virologic response [cEVR]), week 24 or week 48 (end of treatment response [ETR]); the proportion of patients with relapsed viremia (HCV RNA < 15 IU/mL at end of treatment [weeks 24 or 48] that became detectable [≥ 15 IU/mL] at or prior to week 72); and patterns of AEs and safety measures.
Study treatmentsFLV or matching placebo was administered orally at a dose of 300 mg or 600 mg BID. PegIFN (Pegasys, Genentech USA, South San Francisco, CA, USA) was administered at a dose of 180 subcutaneously once weekly. RBV (Copegus, Genentech USA, South San Francisco, CA, USA) was administered at 1,000 mg BID for patients weighing ≤ 75 kg or 1,200 mg BID for patients weighing > 75 kg (given as divided doses). Patients were instructed to take FLV and RBV with food (at least a light meal). The use of clinically significant inhibitors of cytochrome P450 (CYP) 3A4 or sensitive CYP3A4 substrates was not permitted owing to the potential for uncharacterized interactions with FLV. The use of erythropoietin or granulocyte colony-stimulating factors to treat hematologic toxicities associated with pegIFN/RBV therapy was not permitted.
Dose rationaleFLV doses of 300 mg BID and 600 mg BID were selected based on an exposure response analysis of results from a 10-day monotherapy study2 and results from a 4-week study of FLV administered in combination with pegIFN/RBV.3 Predictions from the exposure-response model indicated that exposures associated with the 300 mg BID dose (600 mg total daily dose) would achieve at least half the maximal response seen in the monotherapy studies while exposures associated with the 600 mg BID dose (1,200 mg total daily dose) would likely produce HCV RNA reductions close to the maximal response. Results from the 4-week combination trial demonstrated RVR rates of 60-80% at doses ranging from 200 to 500 mg BID.3
Efficacy analysesPlasma samples were collected for the determination of HCV RNA (COBAS AmpliPrep/COBAS HCV TaqMan assay [research use only; lower limit of detection = 15 IU/mL], Roche, Pleasanton, CA, USA). HCV RNA levels were measured at all study visits (see above). Baseline HCV RNA concentration was defined as the average of the predose values at screening and day 1.
Safety analysesFull physical examinations were conducted at screening, end of treatment, and at final follow-up visits. Targeted physical examinations were conducted at baseline and various timepoints throughout the study. Single 12-lead ECGs were conducted at screening, weeks 2 and 24, and at the end of treatment and final follow-up visits. Vital sign measurements and laboratory safety tests were conducted at all study visits (see above). Laboratory safety test abnormalities were graded per the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events.
Based on observations of adverse testicular effects in nonclinical toxicology studies, an exploratory, randomized, double-blind, placebo-controlled, parallel-group, sub-study of this clinical trial was conducted, evaluating the effect of FLV (300 mg or 600 mg BID) in combination with pegIFN and RBV on semen parameters in treatment-naïve, male HCV genotype 1-infected patients. Parameters evaluated included semen volume, sperm concentration, sperm motility, total sperm count, total motile sperm, sperm pH, and sperm morphology at weeks 12, 24, 36, and 60.
NS5B sequence analysisPlasma samples were collected for NS5B sequence analysis. Baseline samples from all randomized patients were investigated using NS5B population sequencing (Virco BVBA, Beerse, Belgium). Amino acid alignments were generated and used to determine the presence of polymorphisms or substitutions associated with reduced susceptibility to FLV. The analysis focused on the NS5B region encoding the FLV binding site (Thumb 2 domain) and on positions previously identified in preclinical and clinical studies as being associated with reduced susceptibility to FLV.1,2,5 Results presented are from interim/unaudited data.
Statistical methodsThis trial was designed as a Phase 2b estimation study to assess whether the addition of FLV to pegIFN/RBV would significantly increase the proportion of patients achieving SVR at week 72 (primary endpoint). A sample size of 96 patients per arm had ≥ 80% power to detect a 15% increase in the proportion of patients achieving SVR in each of the treatment arms compared with placebo at a one-sided α level of 10%. No multiple comparison adjustment was considered.
The primary analysis included all patients who were randomized and received at least one dose of any study medication. The treatment difference in the proportions of patients achieving SVR, along with the two-sided 80% confidence interval, was constructed adjusting for randomization strata (i.e., screening HCV RNA < 400,000 vs. ≥ 400,000 IU/ mL) using the Cochran-Mantel-Haenszel weights6,7 and the normal approximation to the binomial distribution. Patients with missing HCV RNA values at either the end of treatment or Week 72 were not considered to have achieved SVR.
The safety analysis included all patients who were randomized and received at least one dose of any study medication.
Three interim analyses were conducted in an unblinded manner by the sponsor and the results were used for program decision-making and planning; however, investigators and patients remained blinded until the completion of the Week 24 visit. The results from the final analysis are presented here.
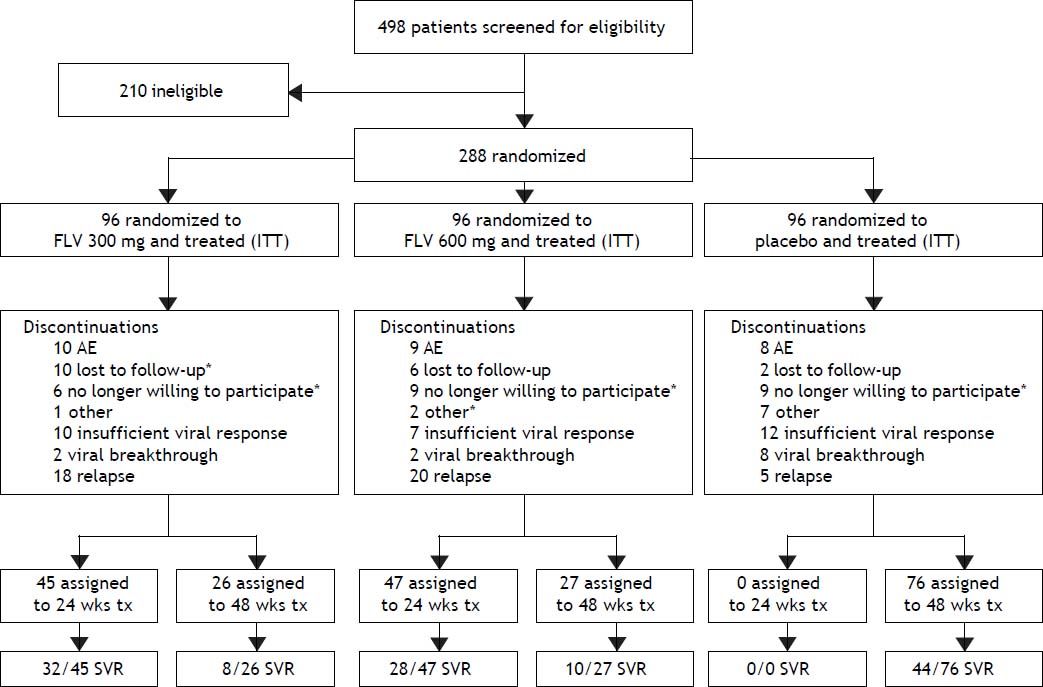
ResultsStudy patients and dispositionA total of 288 patients were randomized and all received at least one dose of study drug and were included in the intent-to-treat (ITT) and safety analyses (Figure 1). Demographic and baseline characteristics were generally well balanced among treatment groups (Table 1). A slight majority of the patients were male (53%) and the majority of the patients were white (79%), with HCV RNA > 400,000 IU/mL at screening (81%), and no or mild fibrosis (83%). The most common reasons for discontinuation (Figure 2) were virologic relapse (n = 43), insufficient virologic response (n = 29), and AEs (n = 27). More patients in the FLV arms discontinued due to virologic relapse compared with the placebo arm. Forty-five and 47 patients in the FLV 300 mg BID and 600 mg BID arms, respectively, met the HCV RNA response-guided criteria and discontinued all therapy at week 24. Patients in the placebo arm were ineligible to discontinue therapy at week 24.
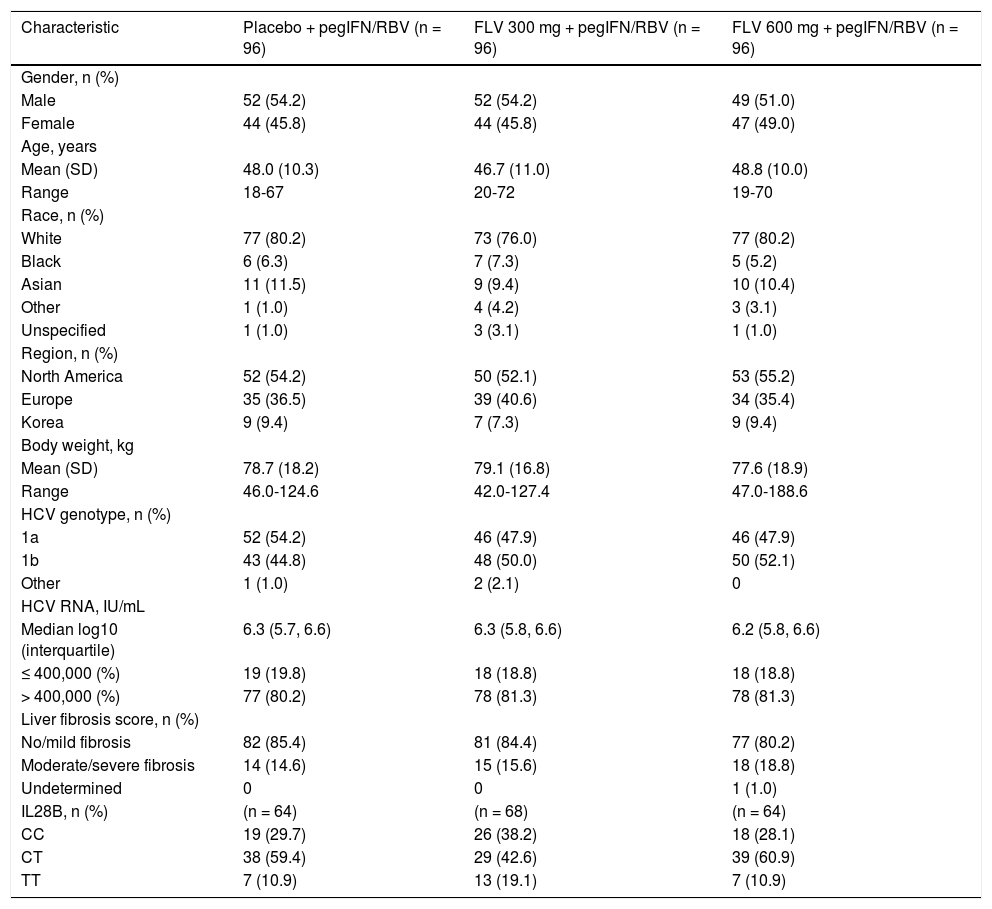
Summary of demographic and baseline characteristics (ITT).
| Characteristic | Placebo + pegIFN/RBV (n = 96) | FLV 300 mg + pegIFN/RBV (n = 96) | FLV 600 mg + pegIFN/RBV (n = 96) |
|---|---|---|---|
| Gender, n (%) | |||
| Male | 52 (54.2) | 52 (54.2) | 49 (51.0) |
| Female | 44 (45.8) | 44 (45.8) | 47 (49.0) |
| Age, years | |||
| Mean (SD) | 48.0 (10.3) | 46.7 (11.0) | 48.8 (10.0) |
| Range | 18-67 | 20-72 | 19-70 |
| Race, n (%) | |||
| White | 77 (80.2) | 73 (76.0) | 77 (80.2) |
| Black | 6 (6.3) | 7 (7.3) | 5 (5.2) |
| Asian | 11 (11.5) | 9 (9.4) | 10 (10.4) |
| Other | 1 (1.0) | 4 (4.2) | 3 (3.1) |
| Unspecified | 1 (1.0) | 3 (3.1) | 1 (1.0) |
| Region, n (%) | |||
| North America | 52 (54.2) | 50 (52.1) | 53 (55.2) |
| Europe | 35 (36.5) | 39 (40.6) | 34 (35.4) |
| Korea | 9 (9.4) | 7 (7.3) | 9 (9.4) |
| Body weight, kg | |||
| Mean (SD) | 78.7 (18.2) | 79.1 (16.8) | 77.6 (18.9) |
| Range | 46.0-124.6 | 42.0-127.4 | 47.0-188.6 |
| HCV genotype, n (%) | |||
| 1a | 52 (54.2) | 46 (47.9) | 46 (47.9) |
| 1b | 43 (44.8) | 48 (50.0) | 50 (52.1) |
| Other | 1 (1.0) | 2 (2.1) | 0 |
| HCV RNA, IU/mL | |||
| Median log10 (interquartile) | 6.3 (5.7, 6.6) | 6.3 (5.8, 6.6) | 6.2 (5.8, 6.6) |
| ≤ 400,000 (%) | 19 (19.8) | 18 (18.8) | 18 (18.8) |
| > 400,000 (%) | 77 (80.2) | 78 (81.3) | 78 (81.3) |
| Liver fibrosis score, n (%) | |||
| No/mild fibrosis | 82 (85.4) | 81 (84.4) | 77 (80.2) |
| Moderate/severe fibrosis | 14 (14.6) | 15 (15.6) | 18 (18.8) |
| Undetermined | 0 | 0 | 1 (1.0) |
| IL28B, n (%) | (n = 64) | (n = 68) | (n = 64) |
| CC | 19 (29.7) | 26 (38.2) | 18 (28.1) |
| CT | 38 (59.4) | 29 (42.6) | 39 (60.9) |
| TT | 7 (10.9) | 13 (19.1) | 7 (10.9) |
CC: cytosine-cytosine. CT: cytosine-thymine. FLV: filibuvir. HCV: hepatitis C virus. IL: interleukin. ITT: intent-to-treat. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin. RNA: ribonucleic acid. SD: standard deviation. TT: thymine-thymine.
 on day 422 which was in the week 72 visit window for analysis. This patient’s EOTR and week 72 HCV RNA were < 15 IU/mL; therefore, the patients was classified as an SVR as per the pre-specified primary analysis. AE: adverse event. EOTR: end of treatment response. HCV: hepatitis C virus. ITT: intent to treat. RNA: ribonucleic acid. SVR: sustained virologic response. tx: treatment. wks: weeks.")
Patient disposition. *One or more discontinuations in this category occurred after virologic relapse. Discontinuations included all patient discontinuations throughout the study. One patient in the FLV300 mg group discontinued (no longer willing to participate) on day 422 which was in the week 72 visit window for analysis. This patient’s EOTR and week 72 HCV RNA were < 15 IU/mL; therefore, the patients was classified as an SVR as per the pre-specified primary analysis. AE: adverse event. EOTR: end of treatment response. HCV: hepatitis C virus. ITT: intent to treat. RNA: ribonucleic acid. SVR: sustained virologic response. tx: treatment. wks: weeks.
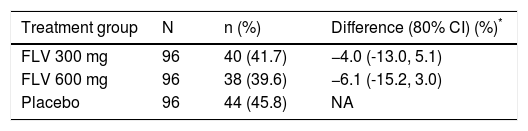
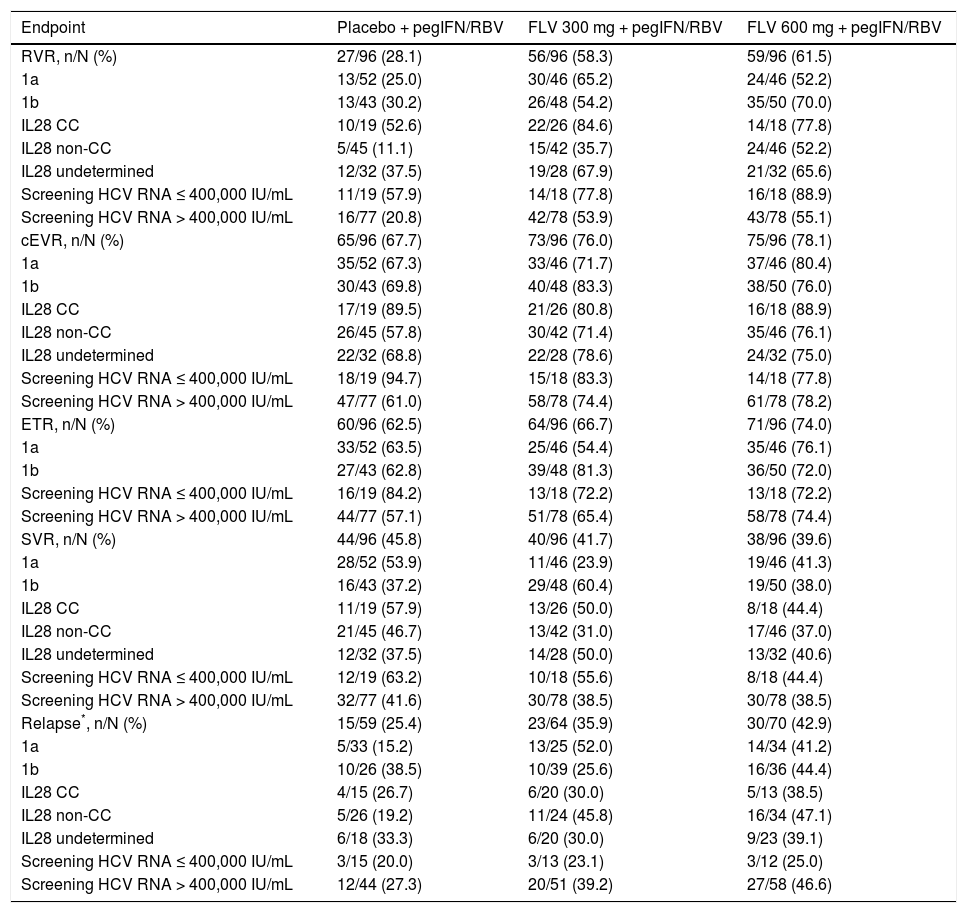
The primary efficacy endpoint for the study was the percentage of patients achieving SVR, and 41.7, 39.6, and 45.8%, of patients in the FLV 300 mg BID, FLV 600 mg BID, and placebo arms achieved this endpoint, respectively (Tables 2 and 3). Thus, the addition of FLV to pegIFN/RBV did not improve the percentage of patients achieving SVR relative to administration of pegIFN/RBV alone in the ITT population (Table 2). Similar results were observed in pre-specified subgroups by HCV genotype 1 subtype (1a vs. 1b), host interleukin (IL)-28 genotype (CC vs. non-CC), and screening HCV RNA level (≤ 400,000 or > 400,000 IU/mL) (Table 3). However, patients in the FLV arms who achieved HCV RNA < 15 IU/mL within the first 2 weeks of therapy experienced higher rates of SVR, despite the fact that the majority of these patients received a shortened duration of therapy and stopped all treatment at week 24 (Table 4). Patients in the FLV arms who discontinued therapy at week 24 had higher rates of SVR (71.1 and 59.6% in the FLV 300 mg BID and 600 mg BID arms, respectively) than patients who received an additional 24 weeks of pegIFN/RBV therapy (30.8 and 37.0% in the FLV 300 mg BID and 600 mg BID arms, respectively) (Table 4). Not achieving HCV RNA < 15 IU/mL within the first 2 weeks of therapy was associated with a lower rate of SVR in all treatment groups, but the impact was more pronounced in the FLV arms.
Primary analysis of percentage of patients with sustained virologic response by treatment group (ITT).
| Treatment group | N | n (%) | Difference (80% CI) (%)* |
|---|---|---|---|
| FLV 300 mg | 96 | 40 (41.7) | −4.0 (-13.0, 5.1) |
| FLV 600 mg | 96 | 38 (39.6) | −6.1 (-15.2, 3.0) |
| Placebo | 96 | 44 (45.8) | NA |
Missing HCV RNA at either end of treatment or week 72 = failure.
Cochran-Mantel-Haenszel estimates adjusted for screening HCV RNA (≤ 400,000 vs. > 400,000 IU/mL). Difference is the percentage difference between the FLV and placebo groups. CI: confidence interval. FLV: filibuvir. HCV: hepatitis C virus. ITT: intent-to-treat. NA: not applicable. RNA: ribonucleic acid.
Primary and secondary endpoints, overall and by key subgroups (ITT).
| Endpoint | Placebo + pegIFN/RBV | FLV 300 mg + pegIFN/RBV | FLV 600 mg + pegIFN/RBV |
|---|---|---|---|
| RVR, n/N (%) | 27/96 (28.1) | 56/96 (58.3) | 59/96 (61.5) |
| 1a | 13/52 (25.0) | 30/46 (65.2) | 24/46 (52.2) |
| 1b | 13/43 (30.2) | 26/48 (54.2) | 35/50 (70.0) |
| IL28 CC | 10/19 (52.6) | 22/26 (84.6) | 14/18 (77.8) |
| IL28 non-CC | 5/45 (11.1) | 15/42 (35.7) | 24/46 (52.2) |
| IL28 undetermined | 12/32 (37.5) | 19/28 (67.9) | 21/32 (65.6) |
| Screening HCV RNA ≤ 400,000 IU/mL | 11/19 (57.9) | 14/18 (77.8) | 16/18 (88.9) |
| Screening HCV RNA > 400,000 IU/mL | 16/77 (20.8) | 42/78 (53.9) | 43/78 (55.1) |
| cEVR, n/N (%) | 65/96 (67.7) | 73/96 (76.0) | 75/96 (78.1) |
| 1a | 35/52 (67.3) | 33/46 (71.7) | 37/46 (80.4) |
| 1b | 30/43 (69.8) | 40/48 (83.3) | 38/50 (76.0) |
| IL28 CC | 17/19 (89.5) | 21/26 (80.8) | 16/18 (88.9) |
| IL28 non-CC | 26/45 (57.8) | 30/42 (71.4) | 35/46 (76.1) |
| IL28 undetermined | 22/32 (68.8) | 22/28 (78.6) | 24/32 (75.0) |
| Screening HCV RNA ≤ 400,000 IU/mL | 18/19 (94.7) | 15/18 (83.3) | 14/18 (77.8) |
| Screening HCV RNA > 400,000 IU/mL | 47/77 (61.0) | 58/78 (74.4) | 61/78 (78.2) |
| ETR, n/N (%) | 60/96 (62.5) | 64/96 (66.7) | 71/96 (74.0) |
| 1a | 33/52 (63.5) | 25/46 (54.4) | 35/46 (76.1) |
| 1b | 27/43 (62.8) | 39/48 (81.3) | 36/50 (72.0) |
| Screening HCV RNA ≤ 400,000 IU/mL | 16/19 (84.2) | 13/18 (72.2) | 13/18 (72.2) |
| Screening HCV RNA > 400,000 IU/mL | 44/77 (57.1) | 51/78 (65.4) | 58/78 (74.4) |
| SVR, n/N (%) | 44/96 (45.8) | 40/96 (41.7) | 38/96 (39.6) |
| 1a | 28/52 (53.9) | 11/46 (23.9) | 19/46 (41.3) |
| 1b | 16/43 (37.2) | 29/48 (60.4) | 19/50 (38.0) |
| IL28 CC | 11/19 (57.9) | 13/26 (50.0) | 8/18 (44.4) |
| IL28 non-CC | 21/45 (46.7) | 13/42 (31.0) | 17/46 (37.0) |
| IL28 undetermined | 12/32 (37.5) | 14/28 (50.0) | 13/32 (40.6) |
| Screening HCV RNA ≤ 400,000 IU/mL | 12/19 (63.2) | 10/18 (55.6) | 8/18 (44.4) |
| Screening HCV RNA > 400,000 IU/mL | 32/77 (41.6) | 30/78 (38.5) | 30/78 (38.5) |
| Relapse*, n/N (%) | 15/59 (25.4) | 23/64 (35.9) | 30/70 (42.9) |
| 1a | 5/33 (15.2) | 13/25 (52.0) | 14/34 (41.2) |
| 1b | 10/26 (38.5) | 10/39 (25.6) | 16/36 (44.4) |
| IL28 CC | 4/15 (26.7) | 6/20 (30.0) | 5/13 (38.5) |
| IL28 non-CC | 5/26 (19.2) | 11/24 (45.8) | 16/34 (47.1) |
| IL28 undetermined | 6/18 (33.3) | 6/20 (30.0) | 9/23 (39.1) |
| Screening HCV RNA ≤ 400,000 IU/mL | 3/15 (20.0) | 3/13 (23.1) | 3/12 (25.0) |
| Screening HCV RNA > 400,000 IU/mL | 12/44 (27.3) | 20/51 (39.2) | 27/58 (46.6) |
Derived based on HCV RNA levels, not investigator reported. Includes only those patients with end of treatment HCV RNA <15 IU/mL. CC: cytosine-cytosine. cEVR: complete early virologic response. ETR: end of treatment response. FLV: filibuvir. HCV: hepatitis C virus. IL: interleukin. ITT: intent-to-treat. pegI-FN: pegylated interferon alfa-2a. RBV: ribavirin. RNA: ribonucleic acid. RVR: rapid virologic response. SVR: sustained virologic response.
Percentage of patients with SVR by time to first HCV RNA < 15 IU/mL and duration of therapy.
| SVR | Placebo + pegIFN/RBV, n/N (%) | FLV 300 mg + pegIFN/RBV, n/N (%) | FLV 600 mg + pegIFN/RBV, n/N (%) |
|---|---|---|---|
| By time to first HCV RNA < 15 IU/mL | |||
| 0-2 weeks | 5/8 (62.5) | 21/32 (65.6) | 21/36 (58.3) |
| > 2-48 weeks | 39/71 (54.9) | 19/48 (39.6) | 17/51 (33.3) |
| Never | 0/17 (0) | 0/16 (0) | 0/9 (0) |
| By duration of therapy* | |||
| 24 weeks | NA | 32/45 (71.1) | 28/47 (59.6) |
| 48 weeks | 44/76 (57.9) | 8/26 (30.8) | 10/27 (37.0) |
Maximum duration of therapy a patient may have received. Patients who discontinued early did not receive a full 24 or 48 weeks of therapy. FLV: filibuvir. HCV: hepatitis C virus. NA: not applicable. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin. RNA: ribonucleic acid. SVR: sustained virologic response.
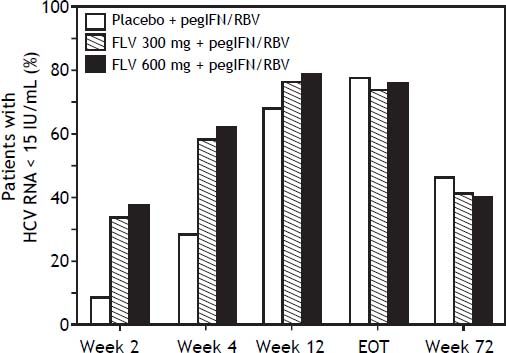
The addition of FLV to pegIFN/RBV improved on-treatment virologic response parameters, such as RVR, cEVR, and ETR (Table 3). Specifically, the percentage of patients with HCV RNA < 15 IU/mL at week 4 (RVR) was numerically higher in the FLV 300 mg BID and 600 mg BID arms (58.3 and 61.5%, respectively) compared with the placebo arm (28.1%), and similar results were observed at week 12 (cEVR; 67.7%, 76.0%, and 78.1% of patients in the placebo, FLV 300 mg BID, and FLV 600 mg BID arms, respectively). While HCV genotype-1 subtype did not result in consistent differences in rates of RVR or cEVR among treatment arms, the negative impact of factors such as IL28 non-CC and high screening HCV RNA levels on rates of RVR and cEVR appeared to be less pronounced in the FLV arms compared with placebo; however, the sample sizes in these subgroups were insufficient to determine the significance of these observations. At the end of treatment (weeks 24 or 48), the percentage of patients with HCV RNA < 15 IU/mL was higher in the FLV 600 mg BID arm compared with the placebo arm, but not in the FLV 300 mg BID arm (Table 3). While the addition of FLV to pegIFN/RBV enabled patients to achieve HCV RNA < 15 IU/mL faster than administration of pegIFN/RBV alone, this did not result in higher rates of SVR (Figure 3; Table 2).
 (intent-to-treat population). Any patient with a missing value at a particular visit was handled as follows: 1) patient was considered not to have undetectable HCV RNA if the patient discontinued from of study before the particular time point; 2) for patients who had not discontinued from the study, LOCF was used. For example, if the HCV RNA vale was missing at week 4, but the patient was still in the study at week 12, the last value prior to week 4 was carried forward for week 4. EOT: end of treatment. FLV: filibuvir. HCV: hepatitis C virus. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin. RNA: ribonucleic acid.")
Proportion of patients with HCV RNA <15 IU/mL (weeks 2,4,12, EOT, week 72) (intent-to-treat population). Any patient with a missing value at a particular visit was handled as follows: 1) patient was considered not to have undetectable HCV RNA if the patient discontinued from of study before the particular time point; 2) for patients who had not discontinued from the study, LOCF was used. For example, if the HCV RNA vale was missing at week 4, but the patient was still in the study at week 12, the last value prior to week 4 was carried forward for week 4. EOT: end of treatment. FLV: filibuvir. HCV: hepatitis C virus. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin. RNA: ribonucleic acid.
The rate of virologic relapse (derived per HCV RNA levels, not investigator defined) in the FLV 300 mg BID and 600 mg BID arms was higher than that observed in the placebo arm (35.9 and 42.9% vs. 25.4%, respectively). This trend was generally consistent across the various subgroups analyzed, with the exception of the subgroup of patients with HCV RNA ≤ 400,000 IU/mL at screening. In this subgroup, the rate of relapse in the FLV 300 mg BID and 600 mg BID arms was similar to that observed in the placebo arm (23.1 and 25.0% vs. 20.0%, respectively); however, the sample sizes in this subgroup were small.
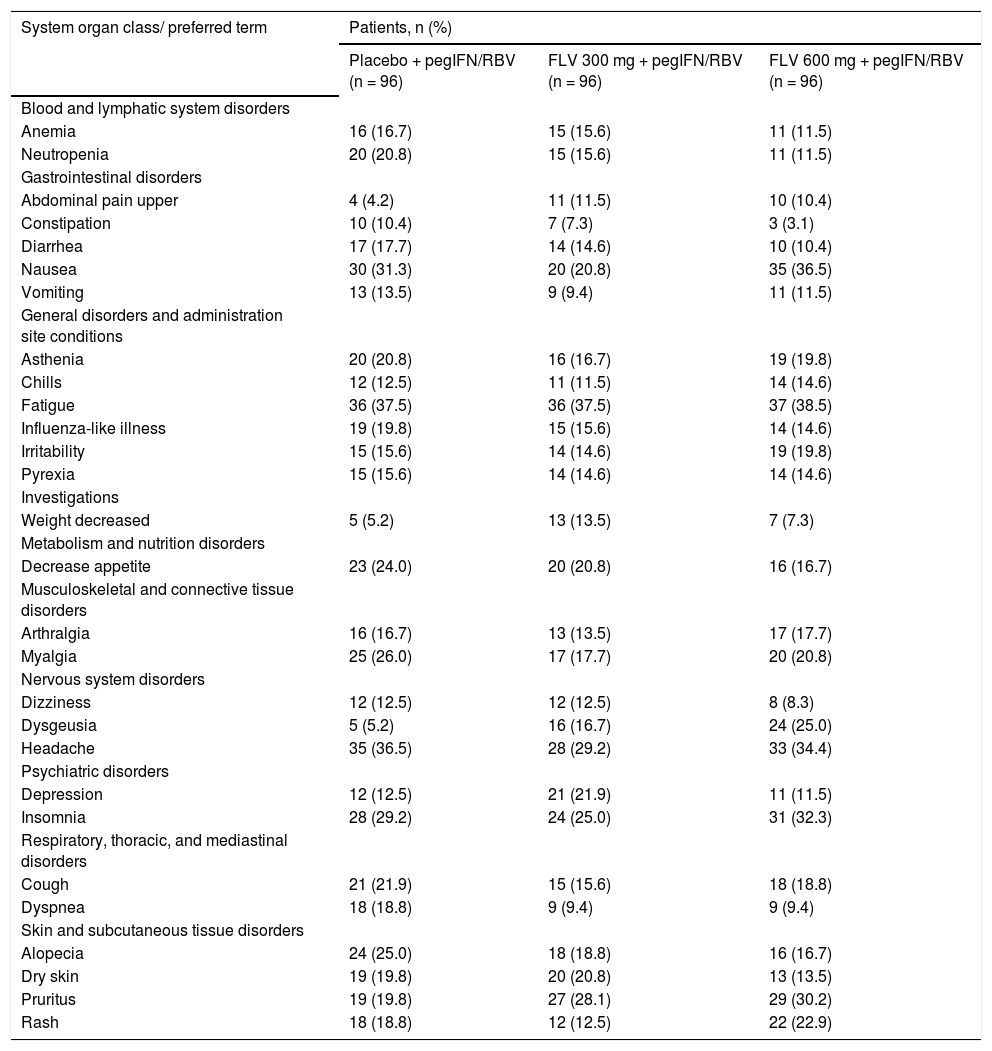
SafetyThe number of patients who discontinued from the study due to AEs was similar across all arms (10, 9, and 8 patients in the FLV 300 mg BID, 600 mg BID, and placebo arms, respectively) (Figure 2). AEs leading to study discontinuation were varied, with no trends identified within or between treatments arms. The most commonly reported AEs were nausea, fatigue, headache, and insomnia, which were reported at similar rates across treatment arms (Table 5). AEs of dysgeusia, upper abdominal pain, and pruritus were more common in the FLV arms than in the placebo arm, and the frequency of dysgeusia and pruritus increased with increasing FLV dose. All reports of dysgeusia and pruritus were mild or moderate in severity and none resulted in premature discontinuation from the study. Most reports of upper abdominal pain were mild or moderate in severity; however, there were two reports of severe abdominal pain (a single case in both the FLV 600 mg and placebo arms), with the patient in the FLV 600 mg discontinuing prematurely from the study. The majority of the remaining AEs were mild or moderate in severity, and severe AEs were reported in similar proportions across treatment arms (data not shown). Twenty-eight patients experienced an SAE (14, 7, and 7 in the FLV 300 mg BID, 600 mg BID, and placebo arms, respectively). There were no deaths in the study.
Treatment-emergent adverse events reported in >10% of patients in any treatment group (all causality).
| System organ class/ preferred term | Patients, n (%) | ||
|---|---|---|---|
| Placebo + pegIFN/RBV (n = 96) | FLV 300 mg + pegIFN/RBV (n = 96) | FLV 600 mg + pegIFN/RBV (n = 96) | |
| Blood and lymphatic system disorders | |||
| Anemia | 16 (16.7) | 15 (15.6) | 11 (11.5) |
| Neutropenia | 20 (20.8) | 15 (15.6) | 11 (11.5) |
| Gastrointestinal disorders | |||
| Abdominal pain upper | 4 (4.2) | 11 (11.5) | 10 (10.4) |
| Constipation | 10 (10.4) | 7 (7.3) | 3 (3.1) |
| Diarrhea | 17 (17.7) | 14 (14.6) | 10 (10.4) |
| Nausea | 30 (31.3) | 20 (20.8) | 35 (36.5) |
| Vomiting | 13 (13.5) | 9 (9.4) | 11 (11.5) |
| General disorders and administration site conditions | |||
| Asthenia | 20 (20.8) | 16 (16.7) | 19 (19.8) |
| Chills | 12 (12.5) | 11 (11.5) | 14 (14.6) |
| Fatigue | 36 (37.5) | 36 (37.5) | 37 (38.5) |
| Influenza-like illness | 19 (19.8) | 15 (15.6) | 14 (14.6) |
| Irritability | 15 (15.6) | 14 (14.6) | 19 (19.8) |
| Pyrexia | 15 (15.6) | 14 (14.6) | 14 (14.6) |
| Investigations | |||
| Weight decreased | 5 (5.2) | 13 (13.5) | 7 (7.3) |
| Metabolism and nutrition disorders | |||
| Decrease appetite | 23 (24.0) | 20 (20.8) | 16 (16.7) |
| Musculoskeletal and connective tissue disorders | |||
| Arthralgia | 16 (16.7) | 13 (13.5) | 17 (17.7) |
| Myalgia | 25 (26.0) | 17 (17.7) | 20 (20.8) |
| Nervous system disorders | |||
| Dizziness | 12 (12.5) | 12 (12.5) | 8 (8.3) |
| Dysgeusia | 5 (5.2) | 16 (16.7) | 24 (25.0) |
| Headache | 35 (36.5) | 28 (29.2) | 33 (34.4) |
| Psychiatric disorders | |||
| Depression | 12 (12.5) | 21 (21.9) | 11 (11.5) |
| Insomnia | 28 (29.2) | 24 (25.0) | 31 (32.3) |
| Respiratory, thoracic, and mediastinal disorders | |||
| Cough | 21 (21.9) | 15 (15.6) | 18 (18.8) |
| Dyspnea | 18 (18.8) | 9 (9.4) | 9 (9.4) |
| Skin and subcutaneous tissue disorders | |||
| Alopecia | 24 (25.0) | 18 (18.8) | 16 (16.7) |
| Dry skin | 19 (19.8) | 20 (20.8) | 13 (13.5) |
| Pruritus | 19 (19.8) | 27 (28.1) | 29 (30.2) |
| Rash | 18 (18.8) | 12 (12.5) | 22 (22.9) |
FLV: filibuvir. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin.
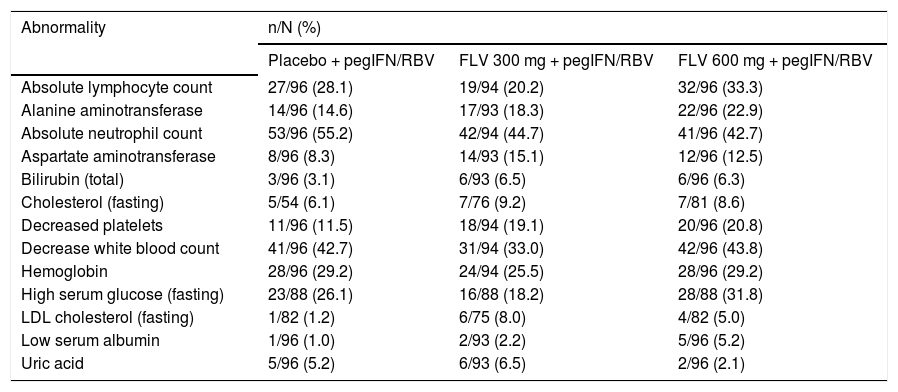
The incidence of laboratory abnormalities grade ≥ 2 was similar across treatment arms (Table 6). The most frequently reported laboratory abnormalities grade ≥ 2 were absolute neutrophil count (44.7, 42.7, and 55.2% in the FLV 300 mg BID, 600 mg BID, and placebo arms, respectively) and decreased white blood cells (33.0, 43.8, and 42.7% in the FLV 300 mg BID, 600 mg BID, and placebo arms, respectively). There was no evidence of a differential effect on semen parameters between treatment arms in the semen sub-study.
Laboratory abnormalities in > 5% of patients in any treatment group (DAIDS grades 2, 3, and 4).
| Abnormality | n/N (%) | ||
|---|---|---|---|
| Placebo + pegIFN/RBV | FLV 300 mg + pegIFN/RBV | FLV 600 mg + pegIFN/RBV | |
| Absolute lymphocyte count | 27/96 (28.1) | 19/94 (20.2) | 32/96 (33.3) |
| Alanine aminotransferase | 14/96 (14.6) | 17/93 (18.3) | 22/96 (22.9) |
| Absolute neutrophil count | 53/96 (55.2) | 42/94 (44.7) | 41/96 (42.7) |
| Aspartate aminotransferase | 8/96 (8.3) | 14/93 (15.1) | 12/96 (12.5) |
| Bilirubin (total) | 3/96 (3.1) | 6/93 (6.5) | 6/96 (6.3) |
| Cholesterol (fasting) | 5/54 (6.1) | 7/76 (9.2) | 7/81 (8.6) |
| Decreased platelets | 11/96 (11.5) | 18/94 (19.1) | 20/96 (20.8) |
| Decrease white blood count | 41/96 (42.7) | 31/94 (33.0) | 42/96 (43.8) |
| Hemoglobin | 28/96 (29.2) | 24/94 (25.5) | 28/96 (29.2) |
| High serum glucose (fasting) | 23/88 (26.1) | 16/88 (18.2) | 28/88 (31.8) |
| LDL cholesterol (fasting) | 1/82 (1.2) | 6/75 (8.0) | 4/82 (5.0) |
| Low serum albumin | 1/96 (1.0) | 2/93 (2.2) | 5/96 (5.2) |
| Uric acid | 5/96 (5.2) | 6/93 (6.5) | 2/96 (2.1) |
DAIDS: Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events. FLV: filibuvir. LDL: low-density lipoprotein. pegIFN: pegylated interferon alfa-2a. RBV: ribavirin.
NS5B sequence analysis was successfully carried out for 268/288 baseline samples. Viruses from 7/268 samples (2.6%) contained NS5B Thumb 2 domain variants associated with >3-fold reduced susceptibility to FLV in vitro (R422K [n = 1]; M423I [n = 1]; M426A/T [n = 1]; V494A [n = 3]; R422K/M426T [n = 1]). The presence of these variants at baseline was not associated with poor on-treatment response to FLV therapy as all affected patients achieved undetectable HCV RNA at weeks 12 and 24. Three of the 7 patients (43%) achieved SVR. While this rate of SVR is similar to that achieved by the overall population, the potential for the presence of baseline mutations to increase the relapse rate cannot be ruled out.
DiscussionThe results of this study demonstrate that while the addition of FLV to pegIFN/RBV produces higher on-treatment virologic response rates compared with administration of pegIFN/RBV alone, a high rate of virologic relapse following discontinuation of therapy resulted in a failure of FLV to improve rates of SVR when administered in combination with pegIFN/RBV as a response-guided regimen.
Studies investigating the use of the HCV protease inhibitors telaprevir and boceprevir demonstrated a relapse rate of ~9% when the products were administered as response-guided regimens.8,9 The expectation at the outset of this study was that the relapse rate among patients treated with FLV in combination with pegIFN/RBV would be similar to that observed for telaprevir and boceprevir. This expectation was based on (1) observations from an earlier FLV clinical trial, where virologic response at week 4 was similar to or better than that reported for telaprevir and boceprevir, respectively,3 and (2) an established association between virologic response at week 4 (RVR) and the likelihood of achieving SVR.10,11 While this association between RVR and SVR was established in trials of pegIFN/RBV, it was anticipated that it would also apply to DAA’s used in combination with pegIFN/RBV. The failure of RVR to translate into high levels of SVR following FLV plus pegIFN/RBV therapy cannot be currently explained. It is known that RBV plays a critical role in reducing relapse in patients undergoing therapy with pegIFN/ RBV; however, an analysis of available pharmacokinetic data suggests no negative interaction between RBV and FLV. The rapid selection of viral variants with reduced susceptibility to FLV may also contribute to the poor correlation between RVR and SVR. Previous studies have demonstrated that single-step mutations are sufficient to confer high-level resistance to FLV;2,5 however, it is not known to what extent this occurs when FLV is administered in combination with pegIFN and RBV.
The relapse rate (35.9-42.9%) observed in the FLV arms of this study is higher than that typically observed with pegIFN/RBV therapy or with other regimens in which a first generation directly-acting antiviral agent is combined with pegIFN/RBV. The high relapse rate observed with FLV therapy may be related to the agent’s potency, genetic barrier to resistance, mechanism of action, or a combination of these characteristics. Relapse/SVR data for other HCV NNIs used in combination with pegIFN/RBV are limited, but the available data suggest a similarly high relapse rate. In a comparable Phase 2 trial, the NNI ANA598 demonstrated a relapse rate of ~25%,12 suggesting a class-related effect.
While FLV administered at doses of 300 mg and 600 mg BID does not improve the SVR rate in HCV-infected patients when combined with pegIFN/RBV, it is possible that this and other such agents may play an important role in future combination antiviral regimens (e.g., IFN-sparing). In fact, the NNI ABT-333 is currently undergoing evaluation in a phase 3 trial as part of an IFN-sparing regimen, and the efficacy associated with this regimen is very compelling.13 While available data would suggest that the NNIs as a class are insufficiently potent to be used in combination with pegIFN/RBV, their use as part of IFN-free combinations represents an optimized role for these agents. Based on the outcome of the present study, the clinical development program for FLV has been suspended; however, several characteristics of FLV, such as its mechanism of action, rapid onset of antiviral activity, consistent activity between genotype 1 subtypes 1a and 1b, and strong safety/tolerability profile, support its evaluation as part of combination antiviral regimens.
Abbreviations- •
AE: adverse event.
- •
BID: twice daily.
- •
cEVR: complete early virologic response.
- •
CYP: cytochrome P450.
- •
ECG: electrocardiogram.
- •
ETR: end of treatment response.
- •
HCV: hepatitis C virus.
- •
IL: interleukin.
- •
ITT: intent-to-treat.
- •
NNI: non-nucleoside inhibitor.
- •
pegIFN: pegylated interferon alfa-2a.
- •
RBV: ribavirin.
- •
RNA: ribonucleic acid.
- •
RVR: rapid virologic response.
- •
SAE: serious AE.
- •
SVR: sustained virologic response.
Maribel Rodriguez-Torres has received grant/research support from Abbott, Akros, Anadys Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, GSK, Idenix Pharmaceuticals, Idera Pharmaceuticals, Inhibitex Pharmaceuticals, Medtronic, Merck, Novartis, Pharmasset, Roche, Santaris Pharmaceuticals, Scynexis Pharmaceuticals, Tibotec, Vertex Pharmaceuticals, ViroChem Pharma, and ZymoGenetics and has served as a consultant for Bristol-Myers Squibb, Genentech, Gilead Sciences, Inhibitex Pharmaceuticals, Janssen, Johnson and Johnson, Merck, and Santaris Pharmaceuticals.
Eric Yoshida has been an investigator of clinical trials sponsored by AbbVie, Boehringer Ingelheim, F. Hoffman-La Roche, Gilead Sciences, Janssen, Merck, Novartis, Pfizer Inc, and Vertex Inc; and has received honoraria for lectures sponsored by F. Hoffman-La Roche Canada, Gilead Sciences Canada, Merck Canada, and Vertex Canada.
Patrick Marcellin has received research grants/ research support from Alios BioPharma, Bristol-Myers Squibb, Gilead Sciences, Janssen-Tibotec, MSD, Novartis, and Roche; has served as a consultant for Abbott, Bristol-Myers Squibb, Gilead Sciences, Janssen-Tibotec, MSD, Novartis, Roche, and Vertex; has served as an investigator for Abbott, Alios BioPharma, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen-Tibotec, MSD, Novartis, Pfizer Inc, Roche, and Vertex; and has served as a speaker for Bristol-Myers Squibb, Gilead Sciences, Janssen-Tibotec, MSD, Novartis, and Roche.
Subasree Srinivasan was employed by Pfizer Inc at the time of the study and holds stock/stock options in Bristol-Myers Squibb and Pfizer Inc.
Vivek S. Purohit is a Pfizer employee.
Cunshan Wang and Jennifer Hammond are Pfizer employees and shareholders.
Financial SupportThis study was sponsored by Pfizer Inc.
AcknowledgmentsThe authors would like to thanks the patients, investigators, nursing staff, and research support staff involved in the study, and the research team at Pfizer Global Research and Development. Editorial support was provided by Karen Irving at Complete Medical Communications and was funded by Pfizer Inc.



