




Background: Hepatitis C virus (HCV) is a major public health problem with 170 million chronically infected people throughout the world. Currently, the only treatment available consists of a combination of pegylated interferon (INF-a) and ribavirin, but only half of the patients treated show a sufficient antiviral response. Thus there is a great need for the development of new treatments for HCV infections. RNA interference (RNAi) represents a new promising approach to develop effective antiviral drugs and has been extremely effective against HCV gene expression in short-term cell culture. Our aim was to determine the effect of RNAi directed against the NS5B-HCV region on HCV expression in a human hepatoma cell line that expresses HCV-subgenomic replicon (Huh7 HCV replicon cells). Methods: We transfected Huh7 HCV replicon cells with different concentrations of RNAi (100-200 nM) targeting the NS5B region of the viral genome. 2-6 days post-transfection HCV-RNA was quantified by semiquantitative and real-time RT-PCR, and HCV NS5B protein levels were assayed by western blot. Cell viability was also quantified by MTT assay. Results: Our results indicate that the NS5B-siRNAs used in this study can specifically inhibit HCV-RNA replication and protein expression (more than 90%) compared to control cells. Conclusions: Synthetic siRNA against NS5B-HCV inhibited HCV replication and viral proteins levels and thereby becomes a powerful strategy to combat hepatitis C virus.
Infection with hepatitis C virus (HCV) is a global public health issue and is a significant cause of cirrhosis, liver failure, hepatocellular carcinoma and liver transplant.1,2 HCV is a member of the Hepacivirus genus in the virus family, Flaviviridae.3 The HCV genome is a single-stranded RNA molecule of positive polarity.4 It contains a single open reading frame (ORF) encoding a polyprotein of about 3,000 amino acids which is cleaved by both cellular and viral proteases to generate 10 functional viral proteins. The structural proteins include the core (C), envelope glycoproteins E1 and E2, and p7. The nonstructural (NS) proteins NS2 to NS5B are involved in polyprotein processing and viral replication.5-7 Particularly, NS5B is the RNA-dependent RNA polymerase (RdRp) that is crucial for viral genome replication.6,7 This viral enzyme has been extensively characterized at the biochemical8 and structural levels9 and has emerged as a major target for antiviral intervention.10,11
Current standard therapy consists of the use of pegylated interferon and the nucleoside analogue ribavirin.12,13 However, this therapy is only effective in 50-60% of infected individuals and is associated with serious side effects,11,13 therefore alternative antiviral drugs that efficiently block virus production are needed.
The search for selective inhibitors of HCV replication has long been hindered by the lack of appropriate cell culture models.14 A major progress was made in 1999 with the generation of HCV replicon-expressing systems, which replicate in human hepatoma cell lines and provide a valuable model to investigate viral expression and for characterizing those factors that regulate HCV expression.15-17 In addition, replicons provide an excellent system to evaluate HCV antiviral agents in cell culture.18
RNAi interference (RNAi) is a cellular process that induces gene silencing.19 RNAi is initiated by the RNase III-like nuclease Dicer, which promotes progressive cleavage of long dsRNAs into 21 to 27 nucleotide (nt) short interfering RNAs (siRNAs) with two nt 3’-over-hangs. Subsequently, the siRNAs are incorporated into an RNA-induced silencing complex (RISC), identified in Drosophila, and the protein-RNA effector nuclease complex recognizes and destroys the target mRNAs.20,21 RNAi has become a powerful and widely used tool for the analysis of gene function and represents a promising new approach to developing effective antiviral drugs.
The HCV genome is a single-stranded RNA that functions as both viral messenger RNA and a template for RNA replication via negative-strand intermediate.4,14 This situation suggests that HCV-RNA molecules could be a particularly attractive target for RNAi therapy with the potential to cure patients infected with HCV. A number of research groups have demonstrated that strong RNAi activity can be induced against HCV using synthetic small interference RNAs (siRNA) duplexes as triggers, or by expressing short hairpin RNAs from plasmid or viral vectors.22-24 However, much work remains to improve delivery, maintain specificity and limit the development of virus resistance. In this study we evaluated the effect of siRNA directed against the RdRp on HCV replicon expression at both RNA and protein level.
Material and methodsCell culture and siRNA transfectionThe original wild-type pFKI389-NS3-3’ replicon DNA from genotype 1b and the generation of Huh7 HCV replicon cells have been described earlier.15 Stable HCV subgenomic replicon cell line was maintained in Dulbecco’s Modified Eagle Medium (DMEM; GIBCO-BRL, Grand Island, NY, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (GIBCO-BRL), 1% (v/v) nonessential amino acids, 100 U of penicillin G per mL and 100 pg of streptomycin per mL. The flasks were maintained in a humidified atmosphere with 5% CO2 at 37°C. The culture medium contained 500 pg/mL of G418 (Geneticin; GIBCO-BRL), which was removed before each experiment. Huh7 HCV replicon cells were incubated with an increasing amount of SIPORT Lipid transfection reagent (Ambion Inc, Austin, TX, USA) (0.3 - 0.6 pL) for up to 48 hours in order to evaluate cell cytotoxicity by tetrazolium salt reduction assay.
Synthetic siRNATo perform RNAi assays two chemically synthesized RNA oligonucleotides (siRNA-NS5B) a directed against the NS5B region of the HCV genome and a siRNA control (non-sense sequence) were designed (Ambion, Inc., Austin, TX, USA). The position and sequence of the synthetic siRNA-NS5B are illustrated in Figure 1. The siR-NA control and siRNA-NS5B (100 and 200 nM) were transfected into Huh7 HCV Replicon cells using SIPORT Lipid reagent for 2, 4 and 6 days. After each experiment total cellular RNA and proteins were isolated from the cell cultures and subjected to RT-PCR semiquantitative and real time RT-PCR to detect HCV-RNA, and immunoblot analysis to identify NS5B viral protein.
RNA Extraction
Total RNA was extracted from Huh7 HCV replicon cells transfected with siRNA-NS5B (100 and 200 nM) or siRNA control using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s specification. RNA was then washed once in 75% alcohol and resuspended in 30 pL of RNase-free water.
RT-PCR for HCV-RNA semiquantificationThe RNA samples were subjected to reverse transcription (RT) using Moloney Murine Leukemia Virus (MMLV) reverse transcriptase and random primers, and the resultant complementary DNAs (cDNAs; 1 pg) were subsequently amplified with Taq DNA polymerase (Promega, Madison, WI, USA) by appropriate PCR cycles (26, 28, 30 and 32 cycles), each one consisting of 2 min at 94°C followed by cycles of 94°C for 1 min, 58°C for 1min and 72°C for 1 min, and finally at 72°C for 10 min. A set of primers, HCV1 (sense; 5’-CTGTGAGGAAC-TACTGTCTTC-3’) and HCV2 (antisense; 5’-CAACAC-TACTCGGCTAGCAGT-3’) were used to amplify a portion of 5’UTR of the HCV genome (221 bp). RT-PCR for Glyceraldehide-3-phosphate dehydrogenase (GAPDH) mRNA was performed in parallel to show an equal amount of total RNA in each sample. The set of primers, gapdh5 (sense; 5’-CCATCACCATCTTCCAGGAGCG - 3’) and gapdh3 (antisense; 5’-AAGGCCATGCCAGT-GAGCTTC -3’) were used for amplify a portion of GAP-DH gene (483 bp). PCR products were visualized in UV light by using a 2% agarose gel.
Real Time RT-PCR for HCV RNA quantificationTotal RNA extracted was subjected to RT using the High-Capacity cDNA Archive Kit (Applied Biosystems) according to the manufacturer’s specification. Two-hundred nanograms of cDNA were subjected to real-time PCR for HCV and GAPDH RNA quantification. Amplifications were conducted in triplicate using the following primers labeled with 6-carboxyfluorescein (6-FAM) and probes labeled with tetrachloro-6-carboxyfluorescein (TAMRA) (Applied Biosystems): HCV replicon TaqMan probe, 5’-6FAM-CTGCACGACACTCATAC-TAMRA-3’; HCV replicon RNA Forward, 5’-GCGTCTAGCCATG-GCGTTA-3’; HCV replicon RNA Reverse, 5’-GGTTCCG-CAGACCACTATGG-3’. Thermal cycling conditions were designed as follows: initial setup at 50°C for 2 min, then 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. For each PCR reaction, 25 pl of TaqMan Universal PCR Master Mix, 2.5 pL of 20X Assay Mix, and 22.5 pL of cDNA diluted in RNase-free water were added. Fluorescence was monitored during every PCR cycle at the annealing step and the amplification plots were generated. GAPDH mRNA expression was used to normalize the RNA concentration in each sample tested. For GAPDH-RNA quantification we used GAPDH (20X) assay (Applied Biosystems: Part number 4326317E) according to the manufacturer’s specification.
Protein extraction and immunoblot assayTotal cell lysates were prepared from Huh7 HCV replicon cells transfected with siRNA-NS5B (100 and 200 nM) or control cells (cells transfected with siRNA control) for 2-6 days. Proteins were extracted with 1X lysis buffer containing 10 mM Tris-HCl pH 7.5, 50 mM KCl, 2 mM MgCl2, 1% Triton X-100, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF) and complete EDTA-free protease inhibitors. After incubation on ice for 20 minutes, the lysates were centrifuged at 13,000 x g for 10 minutes at 4°C. The supernatant was transferred to a fresh tube and protein concentration was measured by the Bradford assay (Bio-Rad). Equal amounts of protein (40 pg) were separated by 10% SDS-PAGE and transferred onto Nitrocellulose membrane (Hybond-ECL Amersham Biosciences). The membranes were blocked with phosphate-buffered saline (PBS)/Triton (0.1% Triton X-100 in PBS, pH 7.2) supplemented with 5% of BSA (w/v) for 1 hour and then incubated overnight at 4°C with one of the following antibodies: goat polyclonal anti-HCV NS5B antibody (dilution 1:500, Santa Cruz Biotechnology, Inc.); goat polyclonal anti-actin antibody (dilution 1:500, Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA). After washing with PBS/Triton, the membranes were incubated with HRP-Rabbit Anti-Goat IgG Conjugate (dilution 1:1000, ZYMED Laboratories, San Francisco, CA, USA) for 2 hours at room temperature. Detection of peroxidase-coupled antibodies was performed with the enhanced chemiluminescence detection system (Roche).
Statistical analysisAll variables were tested in triplicate and experiments were repeated at least three times. One-way analysis of variance was used to test for differences in means and t-test was used for comparisons. The differences were considered significant if P < 0.05.
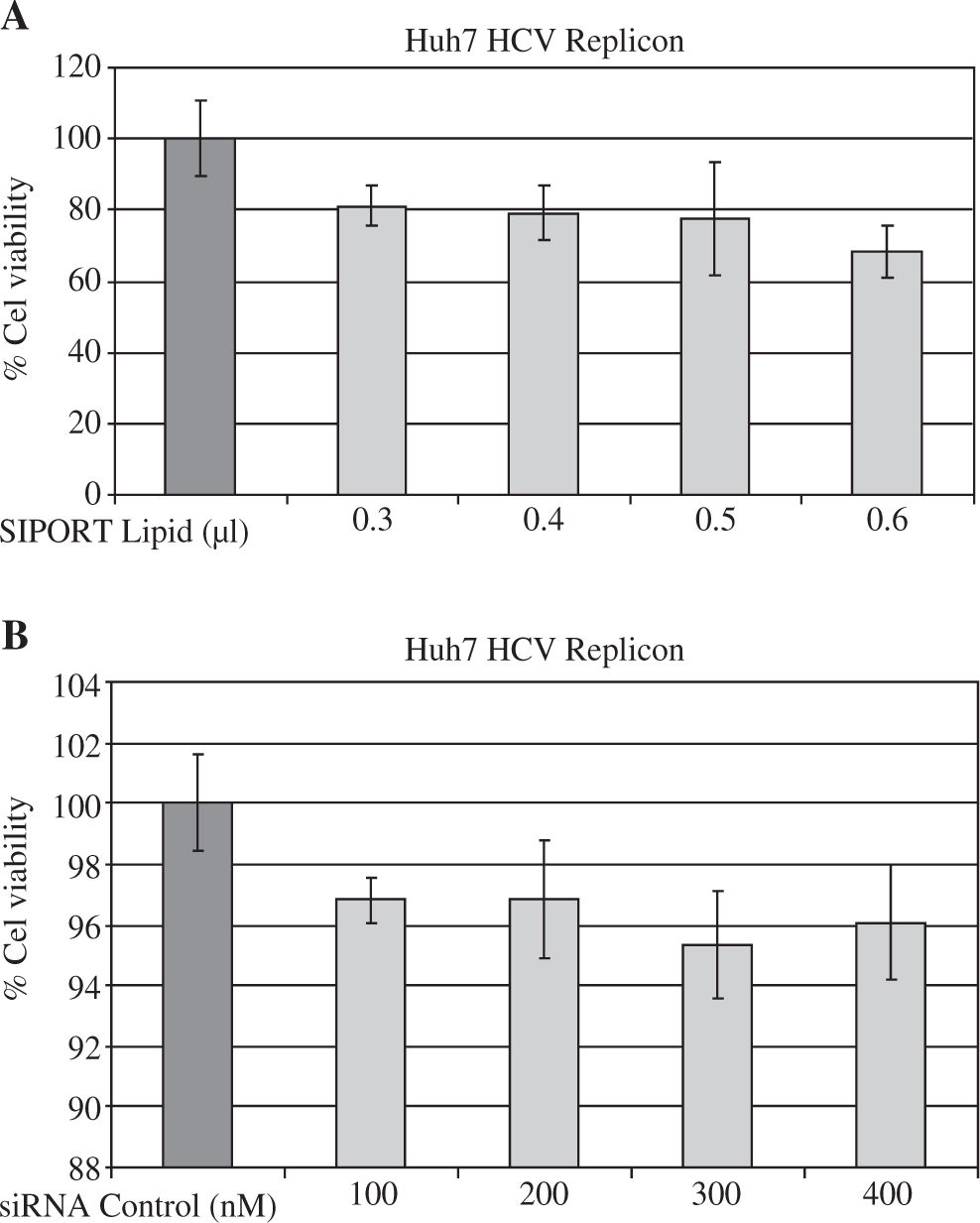
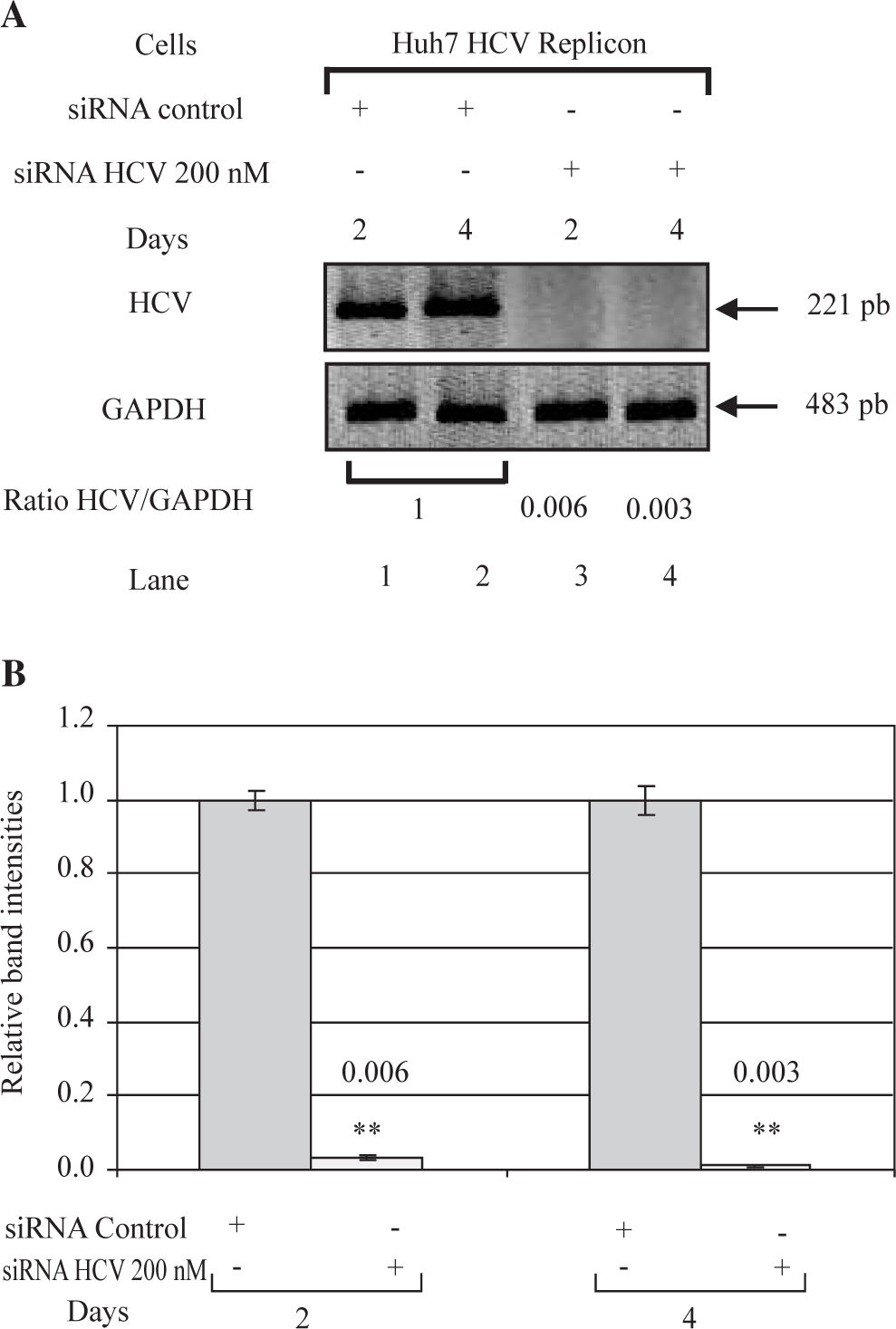
ResultssiRNA-NS5B reduces HCV-RNA levels in Huh7 cells expressing HCV-repliconIn order to determine the right amount of transfection reagent (SIPORT) and siRNA that do not induce cell cytotoxicity, we performed cell viability determinations (MTT assay) using 0.3-0.6pL of the SIPORT reagent and 100-400 nM of siRNA control. We selected the amount of 0.5 pL of transfection reagent (Figure 2A) and 100-200 nM of siRNA which showed no cytotoxic or growth inhibitory effect in culture cells (Figure 2B). To evaluate the effect of siRNA-NS5B (directed against the NS5B region of the viral genome) on HCV-RNA levels, the Huh7 HCV Replicon cells were transfected with 100 and 200 nM of siRNA-NS5B for 2 and 4 days. At the end of each time cells were harvested and HCV-RNA levels were measured by semiquantitative RT-PCR. Semi-quantitative PCR analysis showed that 100 nM of siRNA-NS5B did not appear to have significant effect on HCV-RNA levels (data not shown). However, at a concentration of 200 nM, siR-NA-NS5B dramatically decreased HCV-RNA levels (Figure 3A, lanes 3-4) compared with control cells (cells transfected with siRNA control). The ratio of HCV/GAPDH mRNA from RT-PCR detection was quantified with Phoretix 1D v2003.02 software (Figure 3B).
 The Huh7 HCV replicon cells (5 × 103 cells) plated in a 96-well plate were treated with SI-PORT Lipid transfection reagent at indicated concentrations for 48 hours. (B) The Huh7 HCV replicon cells (5 × 103 cells) plated in a 96-well plate were transfected with 100-400 nM of siRNA control (non-sense sequence) for 48 hours. The cell viability was then determined in triplicates by MTT assay and compared with untreated cells (100% viability).")
Assessing cell viability. (A) The Huh7 HCV replicon cells (5 × 103 cells) plated in a 96-well plate were treated with SI-PORT Lipid transfection reagent at indicated concentrations for 48 hours. (B) The Huh7 HCV replicon cells (5 × 103 cells) plated in a 96-well plate were transfected with 100-400 nM of siRNA control (non-sense sequence) for 48 hours. The cell viability was then determined in triplicates by MTT assay and compared with untreated cells (100% viability).
 Huh7 HCV replicon cells (2 × 105 cells) plated in 6-well plates were transfected with 200 nM siRNA directed against the NS5B region of HCV genome for 2 and 4 days. At the end of incubation total cellular RNA was extracted at each time point and analyzed by semi-quantitative RT-PCR. PCR was performed at 26 cycles. RT-PCR for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was performed in parallel to show an equal amount of total RNA in each sample. (B) The ratio of HCV/GAP-DH mRNA from RT-PCR detection was quantified with Phoretix 1D v2003.02 software. Data are expressed as relative band intensities to control (transfected cells with siRNA control) which is defined as 1.0. The data shown are the mean ± SD of three separate experiments, each one performed in triplicate (**P < 0.01).")
Effect of siRNA on HCV-RNA levels in Huh7 HCV replicon cells. (A) Huh7 HCV replicon cells (2 × 105 cells) plated in 6-well plates were transfected with 200 nM siRNA directed against the NS5B region of HCV genome for 2 and 4 days. At the end of incubation total cellular RNA was extracted at each time point and analyzed by semi-quantitative RT-PCR. PCR was performed at 26 cycles. RT-PCR for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was performed in parallel to show an equal amount of total RNA in each sample. (B) The ratio of HCV/GAP-DH mRNA from RT-PCR detection was quantified with Phoretix 1D v2003.02 software. Data are expressed as relative band intensities to control (transfected cells with siRNA control) which is defined as 1.0. The data shown are the mean ± SD of three separate experiments, each one performed in triplicate (**P < 0.01).
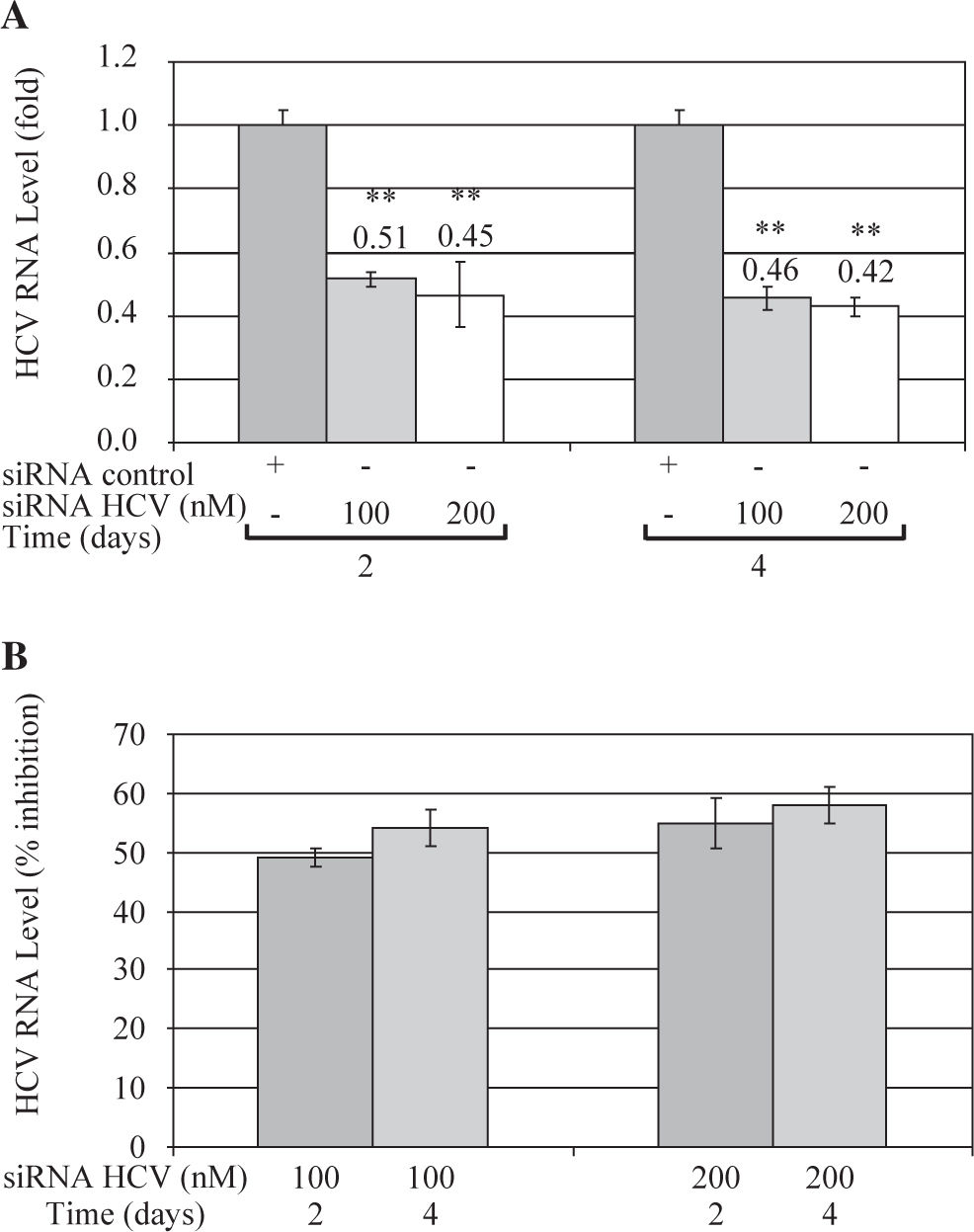
These results were further confirmed by Quantitative Real Time RT-PCR, which is a more sensitive technique. Quantitative analysis showed that both concentrations of siRNA-NS5B (100 and 200 nM) decreased HCV-RNA levels, reaching a maximum inhibition value at 4 days post-transfection (0.46 and 0.42-fold times compared with control cells) (Figure 4A). Figure 4B shows that siRNA-NS5B significantly inhibited HCV RNA level at 2 and 4 days postransfection with both concentrations assayed, reaching a maximum inhibition 4 days postransfection (54% and 58% for 100 and 200 nM, respectively).
 2 × 105 cells plated in 6-well plates were transfected at indicated concentrations of siRNA-NS5B for 2 and 4 days. Viral RNA levels were normalized based on the ratio of HCV/GA-PDH mRNA that was amplified in the same plate by the real-time RT-PCR. Data are expressed as HCV RNA levels relative (fold) to control (transfected cells with siRNA control), which is defined as 1.0. (B) The data are expressed as HCV RNA levels (% inhibition) relative to control that is defined as 0. The data shown are the mean ± SD of triplicate cultures, and the experiment was repeated three times (**P<0.01).")
Quantitative real time RT-PCR of HCV RNA levels from Huh7 HCV replicon cells transfected with 100 and 200nM of siR-NA-NS5B. (A) 2 × 105 cells plated in 6-well plates were transfected at indicated concentrations of siRNA-NS5B for 2 and 4 days. Viral RNA levels were normalized based on the ratio of HCV/GA-PDH mRNA that was amplified in the same plate by the real-time RT-PCR. Data are expressed as HCV RNA levels relative (fold) to control (transfected cells with siRNA control), which is defined as 1.0. (B) The data are expressed as HCV RNA levels (% inhibition) relative to control that is defined as 0. The data shown are the mean ± SD of triplicate cultures, and the experiment was repeated three times (**P<0.01).
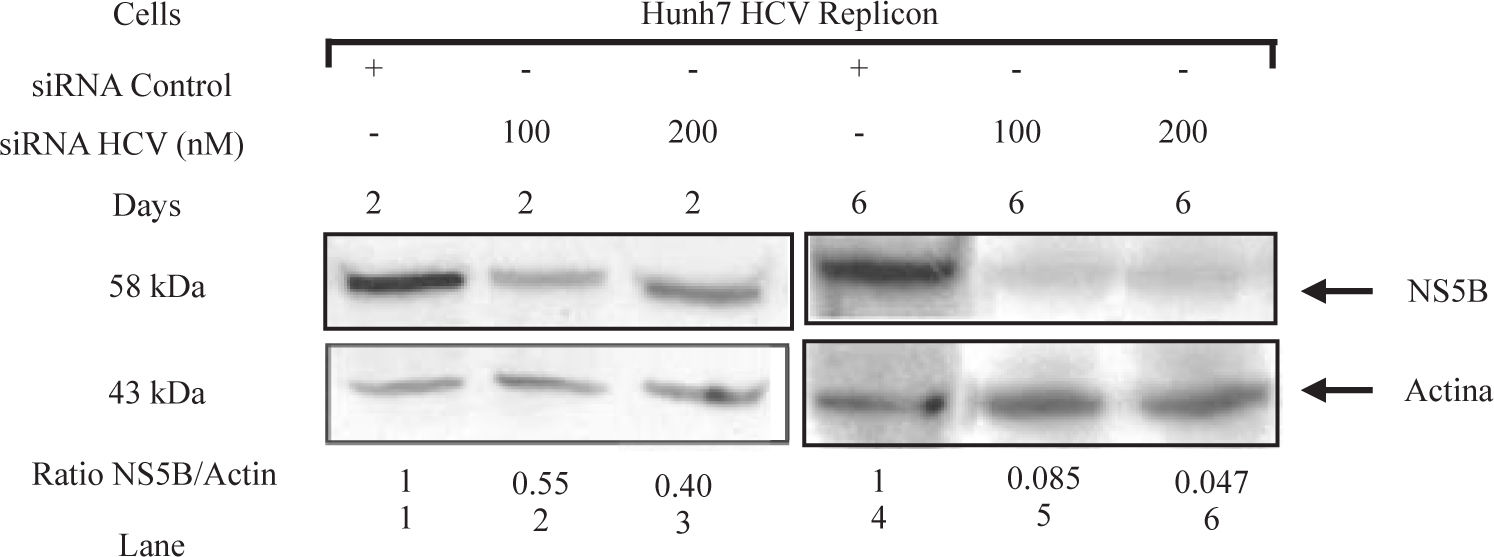
To determine whether siRNAs-NS5B could inhibit the synthesis of viral proteins, NS5B protein levels were determined by Western blot. Replicon cells were transfected with 100 and 200 nM of siRNA-NS5B for up to 6 days and then harvested to perform immunoblot analysis. We observed that siRNA-NS5B at both concentrations used decreased NS5B protein levels several times starting from day 2 until day 6 post-transfection (Figure 5). These data suggest that synthetic siRNA-NS5B not only has a negative effect on HCV-RNA levels, but also it could decrease viral protein production. These results indicate that effect of the siRNA on HCV replicon protein and RNA levels were specific. The effects of siRNA on HCV protein and RNA levels in this study could be the result of siRNA-directed degradation of the HCV replicon RNA by RISC, but at this moment we can not exclude other RNA degradation or decreasing stability mechanisms involved.
 and 200nM of siRNA-NS5B (lanes 3 and 6) for 2 and 6 days. Cell lysates were prepared and equal amounts of protein extracts (40 μg) were subjected to immunoblot analysis to detect NS5B (top panel) and actin levels (bottom panel). The ratio of NS5B/ actin proteins from immunoblot detection was quantified with Phoretix 1D v2003.02 software.")
HCV NS5B protein levels in Huh7 HCV replicon cells transfected with siRNA-NS5B. 2 × 105 Huh7 HCV replicon cells were transfected with 100 nM of siRNA-NS5B (lanes 2 and 5) and 200nM of siRNA-NS5B (lanes 3 and 6) for 2 and 6 days. Cell lysates were prepared and equal amounts of protein extracts (40 μg) were subjected to immunoblot analysis to detect NS5B (top panel) and actin levels (bottom panel). The ratio of NS5B/ actin proteins from immunoblot detection was quantified with Phoretix 1D v2003.02 software.
HCV is an RNA virus and, in many cases, is difficult to eradicate from infected individuals even with an intensive antiviral therapy that utilizes pegylated interferon alfa and ribavirin.25,26 Although a number of other antiviral compounds, including inhibitors against the NS3-4A protease12,27,28 and NS5B RNA dependent RNA polymerase,11,27,29,30 are currently being tested for their therapeutic applicability, such attempts have not always been promising.
Synthetic siRNAs have been used both in vitro and in vivo to cause gene silencing.31,32 The potential of using RNAi activity for treatment of viral diseases and cancer has aroused a great deal of interest in the scientific community. The mechanisms of gene silencing by RNA interference does not induce non-specific intracellular interferon defense mechanisms and therefore represents a promising approach to elucidate gene function or to inhibit certain RNA viruses such as HCV.33
The HCV genome is a positive-sense single-stranded RNA that functions as both a messenger RNA and replication template via a negative-strand intermediate, making it an attractive target for the study of RNA interference. A number of groups have demonstrated that siRNAs interfere with HCV gene expression and replication.22,23 Other laboratories have reported the use of siRNA against HIV-1, HPV and poliovirus in culture cells.34,35 We have demonstrated that introduction of siRNA-NS5B into target cells that contained HCV replicon caused a dramatic decrease of viral RNA and protein levels (Figure 4 and 5). This effect was likely due to the degradation of HCV messenger RNA by the RISC endonuclease. We do not know the effect of RNAi on HCV immediately after virus entry into cells because an efficient cell-culture system for growth of HCV is not available at this time. Because of the great variability in RNA sequences between different quasispecies and genotypes of HCV, for therapeutic applications it may be necessary to include several different combinations of siRNA to target a particular region of the genome. In addition, the high mutation rate of HCV that is apparent during replication makes the appearance of escape mutant from siR-NA. The utility of siRNA as a therapy against HCV infection will depend on the development of efficient delivery systems that induce long-lasting RNAi activity. HCV is an attractive target because of its localization in the infected liver, an organ that can be readily targeted by nucleic acid molecules and viral vectors. In the future, chemically modified synthetic siRNAs, with improved resistance to nucleases coupled with enhanced duration of RNAi, may become a possibility for therapeutic applications. On the other hand, gene therapy offers another possibility to express siRNAs that target HCV in a patient’s liver. The data in this study suggest that siRNAs targeting NS5B viral poly-merase can elicit an anti-HCV response in cell culture. It represents a promising therapy that could eliminate viral RNA from the infected cell and potentially cure patients with hepatitis C.
AcknowledgmentsThis work was supported by grants from the SEP, Mexico Grant Number: PROMEP/103.5/04/2590 to AMR and by the Autonomous University of Nuevo Leon Grant Number: PAICYT SA1010-04 to AMR and SA1436-06 to LTA.



