



Introduction. Balapiravir (R1626, RG1626) is the prodrug of a nucleoside analogue inhibitor of the hepatitis C virus (HCV) RNA-dependent RNA polymerase (R1479, RG1479). This phase 2, double-blind international trial evaluated the optimal treatment regimen of balapiravir plus peginterferon alfa-2a (40KD)/ribavirin.
Material and methods. Treatment-naive genotype 1 patients (N = 516) were randomized to one of seven treatment groups in which they received balapiravir 500, 1,000, or 1,500 mg twice daily, peginterferon alfa2a (40KD) 180 or 90 Mg/week and ribavirin 1,000/1,200 mg/day or peginterferon alfa-2a (40KD)/ribavirin. The planned treatment duration with balapiravir was reduced from 24 to 12 weeks due to safety concerns.
Results. The percentage of patients with undetectable HCV RNA was consistently higher in all balapiravir groups from week 2 to 12. However, high rates of dose modifications and discontinuations of one/all study drugs compromised the efficacy assessment and resulted in similar sustained virological response rates in the balapiravir groups (range 32-50%) and the peginterferon alfa-2a (40KD)/ribavirin group (43%). Balapiravir was discontinued for safety reasons in 28-36% of patients (most often for lymphopenia) and the percentage of patients with serious adverse events (especially hematological, infection, ocular events) was dose related. Serious hematological adverse events (particularly neutropenia, lymphopenia) were more common in balapiravir recipients. Two deaths in the balapiravir/peginterferon alfa-2a/ribavirin combination groups were considered possibly related to study medication.
Conclusion. Further development of balapiravir for the treatment of chronic hepatitis C has been halted because of the unacceptable benefit to risk ratio revealed in this study (www.ClinicalTrials.gov NCT 00517439).
Hepatitis C virus (HCV) infection is the leading cause of liver disease worldwide with an estimated prevalence of more than 170 million individuals.1 Most infected persons develop chronic disease that can progress to cirrhosis, hepatocellular carcinoma, and liver failure. As a result, chronic hepatitis C is the leading cause of liver transplantation in the US.2 Treatments are available for chronic hepatitis C; however, the current standard of care, pegylated interferon plus ribavirin, is most effective against HCV genotype 2 or 3 infection and less effective against the more prevalent genotype 1 infection.3 Several classes of direct-acting antiviral agents have been discovered and are being evaluated in human clinical trials with the goal of increasing cure rates for chronic hepatitis C.
Balapiravir (RG1626) is an orally administered tri-isobutyl ester prodrug of a potent nucleoside analogue inhibitor of the RNA-dependent RNA polymerase (RdRp) of HCV (RG1479).4,5 When administered alone for 14 days at dosages ranging from 1,000 mg/day to 9,000 mg/day, balapiravir produced mean reductions in serum HCV RNA levels up to 3.7 log10 with an acceptable safety profile.6 When administered for 28 days at a dosage of 3 g/day in combination with peginterferon alfa-2a (40 KD) plus ribavirin, the rapid virological response (RVR) rate (HCV RNA <15 IU/mL at week 4) was 74%.7 Importantly, balapiravir-resistant HCV variants were not detected during these short-term clinical trials.6,7 Dose-dependent hematological abnormalities were observed with the triple therapy regimen; however, these events were effectively managed with dose reductions and were reversible upon discontinuation of balapiravir. As a result of these promising results a phase 2 clinical trial was initiated to identify the optimal treatment regimen of balapiravir in combination with peginterferon alfa-2a (40KD) and ribavirin in treatment-naive genotype 1-infected individuals.
Materials and MethodsPatientsAdult patients aged 18 to 65 years with HCV genotype 1 infection who had never received treatment for chronic hepatitis C were eligible for the trial. Chronic hepatitis C was defined as the presence of anti-HCV antibodies and an HCV RNA titer ≥ 50,000 IU/mL in serum (COBAS® Ampliprep/COBAS® TaqMan® HCV test; detection limit 15 IU/mL, Roche Diagnostics, Indianapolis, USA) with a liver biopsy obtained within the previous 24 months (36 months in patients with cirrhosis or incomplete/ transition to cirrhosis) consistent with chronic hepatitis C. HCV genotype 1 infection was confirmed by a molecular assay (Versant HCV Genotyping 2.0 Assay [LiPA], Bayer Diagnostics And Innogenetics, NY, USA).
Patients with advanced fibrosis according to a biopsy obtained within the previous 36 months were required to have compensated liver disease (ChildPugh grade A), a serum α-fetoprotein level < 100 ng/mL, and no evidence of hepatocellular carcinoma on an ultrasound, computerized tomography, or magnetic resonance imaging scan performed within the previous 2 months.
Patients were not eligible if they were infected with any HCV genotype other than genotype 1 or had serological evidence of infection with hepatitis B virus or human immunodeficiency virus. Patients were also excluded if they had a body mass index < 18 kg/m2 or > 36 kg/m2, an absolute neutrophil count < 2 × 109 cells/L, a platelet count < 90 × 109 cells/L, a hemoglobin concentration < 120 g/L in women or < 130 g/L in men (or in patients with risk factors for anemia or in whom anemia would be medically problematic), or a serum creatinine level > 1.5 times the upper limit of normal. Use of erythropoietin-stimulating agents or colony stimulating factors to elevate hematology parameters to facilitate entry into the study was prohibited. Patients who had previously received any interferon preparation, ribavirin (or ribavirin analog), or any investigational HCV protease or polymerase inhibitor were excluded, as were those with a history or evidence of a chronic liver disease other than chronic hepatitis C, a current or past history of chronic disease (including severe psychiatric or pulmonary disease), or a history or evidence of a clinically relevant ophthalmological disorder (e.g. cytomegalovirus infection or macular degeneration). Pregnant or breast-feeding females and male partners of pregnant females were ineligible for the trial.
Female patients of childbearing potential and male patients with partners of childbearing potential were required to use two forms of effective contraception during treatment and after the last dose of ribavirin in accordance with the locally approved label for ribavirin.
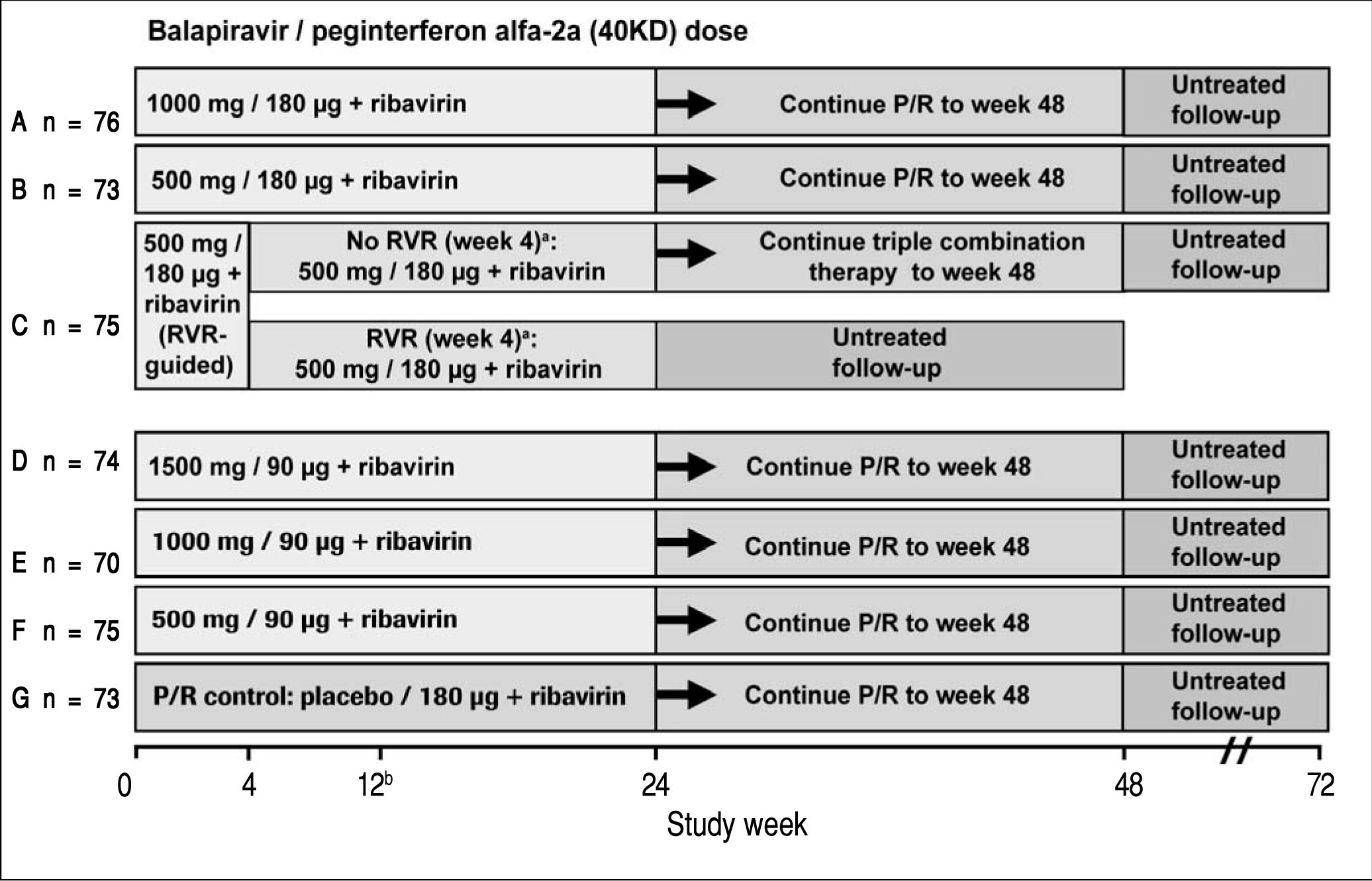
Study designPatients eligible for this phase 2 double-blind, active-controlled, parallel-group, international trial were randomized equally (stratified by geographic region) to one of seven treatment groups (Figure 1). Patients in six groups (A to F) were assigned to receive oral balapiravir (as 500 mg tablets) at a dosage of 500 mg, 1,000 mg, or 1,500 mg twice daily (bid) in combination with subcutaneous peginterferon alfa-2a (40KD) (PEGASYS®, Roche, Basel, Switzerland) at a dosage of 180 μg or 90 μg once weekly and oral ribavirin (COPEGUS® as 200 mg tablets, Roche, Basel, Switzerland) at a dosage of 1,000 mg/day (body weight < 75 kg) or 1,200 mg/day (body weight ≥ 75 kg). Patients in groups A, B, D, E, and F were to stop balapiravir at week 24 and continue with peginterferon alfa-2a (40KD) 180 μg/ week plus ribavirin 1,000/1,200 mg/day for a further 24 weeks to complete a total treatment duration of 48 weeks. The duration of treatment for patients in group C was based on the week 4 HCV RNA assay result: those patients with undetectable HCV RNA in serum (< 15 IU/mL) at week 4 and who remained HCV RNA undetectable through week 22 were to stop all treatment at week 24; those patients who did not meet this criterion were to continue the three-drug combination for a further 24 weeks to complete a total treatment duration of 48 weeks.
 plus ribavirin from 24 to 36 weeks. Patients that had completed more than 12 weeks of treatment with balapiravir were required to stop balapiravir immediately and switch to peginterferon alfa-2a (40KD) plus ribavirin to complete 48 weeks of treatment. Balapiravir was given twice daily (bid). Peginterferon alfa-2a (40KD) was given once weekly. Ribavirin was given at a dosage of 1,000 mg/day to patients weighing < 75 kg and 1,200 mg/day to patients weighing ≥75 kg. The daily dosage of ribavirin was divided into two, each half was administered bid with food. RVR: undetectable HCV RNA (< 15 IU/mL) in serum at week 4. P/R: Peginterferon alfa-2a (40KD) 180 μg/week + ribavirin 1,000/1,200 mg/day.")
Study design including the planned number of patients in each treatment group. aTherapy was stopped at week 24 in patients with an RVR in group C, provided that their HCV RNA had remained undetectable at all visits through to week 22. bA protocol amendment on May 29, 2008 specified that treatment with balapiravir was to be stopped immediately in all patients in group D, and that treatment with balapiravir was to be stopped at week 12 rather than week 24 in all other treatment groups with a corresponding increase in the length of treatment with peginterferon alfa-2a (40KD) plus ribavirin from 24 to 36 weeks. Patients that had completed more than 12 weeks of treatment with balapiravir were required to stop balapiravir immediately and switch to peginterferon alfa-2a (40KD) plus ribavirin to complete 48 weeks of treatment. Balapiravir was given twice daily (bid). Peginterferon alfa-2a (40KD) was given once weekly. Ribavirin was given at a dosage of 1,000 mg/day to patients weighing < 75 kg and 1,200 mg/day to patients weighing ≥75 kg. The daily dosage of ribavirin was divided into two, each half was administered bid with food. RVR: undetectable HCV RNA (< 15 IU/mL) in serum at week 4. P/R: Peginterferon alfa-2a (40KD) 180 μg/week + ribavirin 1,000/1,200 mg/day.
Patients assigned to the control group (G) received oral balapiravir placebo (up to week 24) in combination with peginterferon alfa-2a (40KD) plus ribavirin for 48 weeks.
Double-blinding of the dosage of balapiravir and peginterferon alfa-2a (40KD) was maintained throughout the experimental portion of the study (week 0-24). Blinding of the balapiravir dosage was maintained by providing three bottles of tablets containing balapiravir or placebo and having patients take one tablet from each bottle bid (patients in group G received only matching placebo tablets). Dose reductions were achieved by reducing the number of tablets administered according to a predefined algorithm. Blinding of the dosage of peginterferon alfa-2a (40KD) was maintained by the administration of a similar initial volume of solution (1.0 mL) in all patients and by achieving dose reductions through changes in the volume administered.
Physicians were permitted to reduce the dosage of any study drug in a step-wise manner in order to manage laboratory abnormalities or adverse events. The dosage of balapiravir was reduced in 500 mg/ day (1 tablet) decrements. The dosage of peginterferon alfa-2a (40KD) was reduced in 0.25 mL/week decrements (i.e. from a weekly dose of 180 μg to 135 μg, 90 μg, and 45 μg weekly in patients assigned to 180 μg/week, and from a weekly dose of 90 μg to 68 μg, 45 μg, and 23 μg weekly in patients assigned to 90 μg/week). The dosage of ribavirin was reduced in 200 mg/day (1 tablet) decrements. Investigators were allowed to increase the dosage of study drug after the resolution or an improvement of the precipitating event; however, in the case of balapiravir, escalation to the full starting dose was not recommended. Dose modification schedules for balapiravir and peginterferon alfa-2a (40KD) were included in the protocol for reductions in neutrophil counts, platelet counts and lymphocyte counts, and for balapiravir and ribavirin for reductions in the hemoglobin concentration. Treatment with balapiravir and peginterferon alfa-2a (40KD) was withheld in the event of a reduction in the neutrophil count to < 0.5 cells × 109/L, or a reduction in the platelet count to < 25 cells × 109/L. Treatment with balapiravir was to be permanently discontinued in the event of a reduction in the lymphocyte count to < 0.5 cells × 109/L. Treatment with balapiravir and ribavirin was withheld in the event of a reduction in the hemoglobin concentration to < 85g/L. Off-label use of erythropoietin-stimulating agents and granulocyte colony stimulating factors to manage hematological adverse events or laboratory abnormalities was neither recommended nor prohibited in the protocol.
Protocol amendmentsThe protocol was amended for safety reasons on May 29, 2008 after 516 patients (100%) were enrolled. All patients in group D stopped treatment with balapiravir and switched to open-label treatment with peginterferon alfa-2a (40KD) plus ribavirin. The planned duration of treatment with balapiravir (or balapiravir placebo) was decreased from 24 weeks to 12 weeks in all other treatment groups. Patients who had completed 12 or more weeks of treatment with balapiravir in any group were required to stop balapiravir and switch to open-label treatment with peginterferon alfa-2a (40KD) plus ribavirin. The planned duration of treatment (24 or 48 weeks in group C and 48 weeks in all other groups) was unchanged and was to be achieved by extending the duration of treatment with peginterferon alfa-2a (40KD) plus ribavirin.
The protocol was subsequently amended on October 29, 2008 because of reports of persistent lymphopenia. All patients were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peginterferon alfa-2a (40KD) and ribavirin was permanently discontinued. Among those with CD4+ cell counts < 500 cells/mm3 frequent monitoring for opportunistic infections was required and consideration of prophylactic treatment for opportunistic infections was recommended.
Study conductThe study was conducted in accordance with the Declaration of Helsinki and the laws of the countries in which the study was conducted. The protocol was reviewed by institutional review boards at study sites in the US and by Independent Ethics Committees at study sites in the European Union/ European Economic Area. Each patient provided informed written consent prior to participating in the study (www.ClinicalTrials.gov NCT 00517439).
A data monitoring committee conducted three scheduled interim safety reviews after approximately 175 patients reached treatment weeks 4, 8, and 12, and additional ad hoc reviews when all patients completed study weeks 12 and 24.
The computerized randomization list was generated by the sponsor, maintained in a central repository accessible only to the randomization list managers, and incorporated in double-blind labelling of medication containers. Study site staff were required to call an interactive voice response system and enter a patient number at the time of randomization; at week 24, study sites were to call the interactive voice response system to receive specific instructions on whether to continue treatment with peginterferon alfa-2a (40KD) plus ribavirin or to stop treatment. With the exception of patients in group C, investigators and patients remained blinded to each individual patient’s treatment assignment throughout the study.
EndpointsHCV RNA was determined at baseline and at weeks 1, 2, 3, 4, 6, 8, 12, 18, 22, 24, 30, 36, 42, and 48 during treatment and at weeks 4, 12, and 24 of untreated follow-up.
The primary efficacy outcome of the trial was sustained virological response (SVR), defined as undetectable HCV RNA (< 15 IU/mL) after 24 weeks of untreated follow-up (study week 48 in patients with an RVR in group C and study week 72 in all other patients). The number of patients with an RVR at week 4 and a complete early virological response (complete EVR, undetectable HCV RNA) at week 12 were secondary outcomes.
Patients were required to stop treatment for insufficient therapeutic response if they did not have an EVR after 12 weeks of treatment, defined as undetectable HCV RNA in serum (< 15 IU/mL) or a ≥ 2 log10 drop in HCV RNA titer.
Statistical considerationsThe planned enrolment was 490 patients (70 per group). Mean virological response rates and their corresponding 95% confidence intervals were calculated for each visit for each treatment group. No formal pair-wise statistical comparisons were specified in the protocol. All efficacy analyses were conducted according to the intention to treat principle (all randomized patients who received at least one dose of study drug were included).
ResultsThe first patient was enrolled on November 11, 2007 and the last patient visit occurred on August 4, 2009. A total of 516 patients from 64 centers in Australia, Canada, France, Germany, Italy, Spain and the USA were randomized, and 504 received at least one dose of study medication.
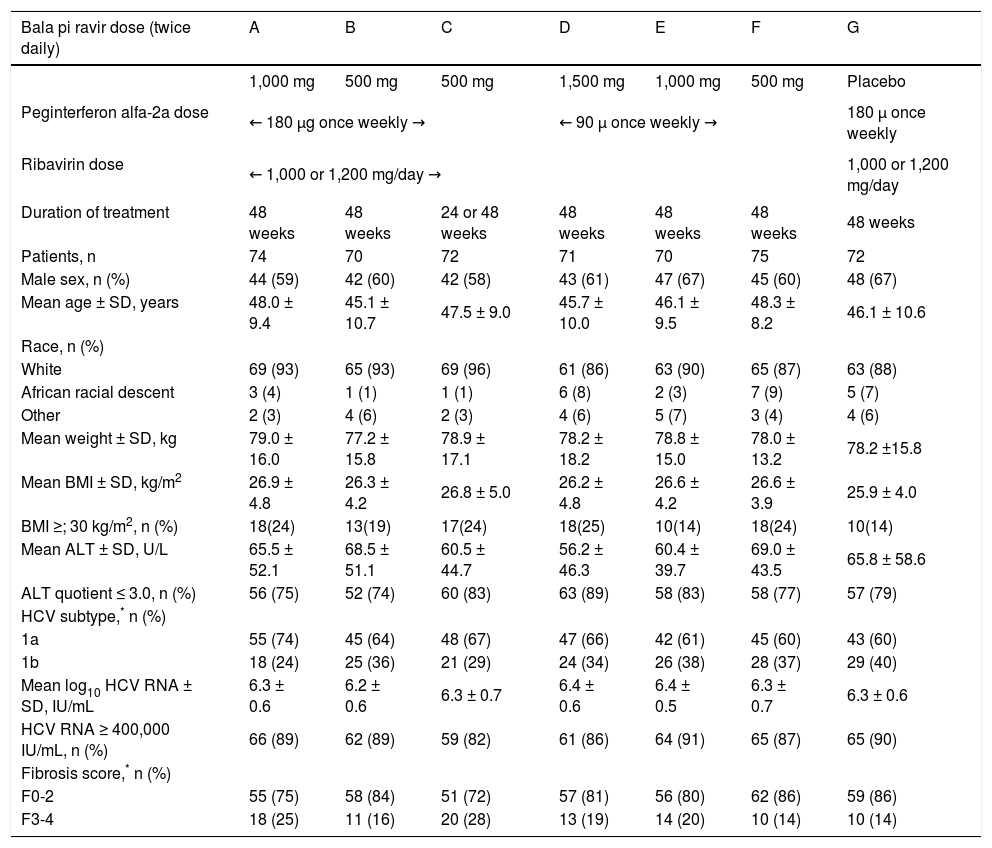
The majority of randomized patients were male (58 to 67%), White (86 to 96%) and infected with HCV genotype 1a (60 to 74%) (Table 1). The mean age ranged from 45 to 48 years, the mean weight ranged from 77 kg to 79 kg and the percentage of patients with advanced fibrosis on the pretreatment liver biopsy ranged from 14 to 28% (Table 1).
Baseline characteristics of patients receiving study drug into the seven treatment groups (A-G) in the current study.
| Bala pi ravir dose (twice daily) | A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|---|
| 1,000 mg | 500 mg | 500 mg | 1,500 mg | 1,000 mg | 500 mg | Placebo | |
| Peginterferon alfa-2a dose | ← 180 μg once weekly → | ← 90 μ once weekly → | 180 μ once weekly | ||||
| Ribavirin dose | ← 1,000 or 1,200 mg/day → | 1,000 or 1,200 mg/day | |||||
| Duration of treatment | 48 weeks | 48 weeks | 24 or 48 weeks | 48 weeks | 48 weeks | 48 weeks | 48 weeks |
| Patients, n | 74 | 70 | 72 | 71 | 70 | 75 | 72 |
| Male sex, n (%) | 44 (59) | 42 (60) | 42 (58) | 43 (61) | 47 (67) | 45 (60) | 48 (67) |
| Mean age ± SD, years | 48.0 ± 9.4 | 45.1 ± 10.7 | 47.5 ± 9.0 | 45.7 ± 10.0 | 46.1 ± 9.5 | 48.3 ± 8.2 | 46.1 ± 10.6 |
| Race, n (%) | |||||||
| White | 69 (93) | 65 (93) | 69 (96) | 61 (86) | 63 (90) | 65 (87) | 63 (88) |
| African racial descent | 3 (4) | 1 (1) | 1 (1) | 6 (8) | 2 (3) | 7 (9) | 5 (7) |
| Other | 2 (3) | 4 (6) | 2 (3) | 4 (6) | 5 (7) | 3 (4) | 4 (6) |
| Mean weight ± SD, kg | 79.0 ± 16.0 | 77.2 ± 15.8 | 78.9 ± 17.1 | 78.2 ± 18.2 | 78.8 ± 15.0 | 78.0 ± 13.2 | 78.2 ±15.8 |
| Mean BMI ± SD, kg/m2 | 26.9 ± 4.8 | 26.3 ± 4.2 | 26.8 ± 5.0 | 26.2 ± 4.8 | 26.6 ± 4.2 | 26.6 ± 3.9 | 25.9 ± 4.0 |
| BMI ≥; 30 kg/m2, n (%) | 18(24) | 13(19) | 17(24) | 18(25) | 10(14) | 18(24) | 10(14) |
| Mean ALT ± SD, U/L | 65.5 ± 52.1 | 68.5 ± 51.1 | 60.5 ± 44.7 | 56.2 ± 46.3 | 60.4 ± 39.7 | 69.0 ± 43.5 | 65.8 ± 58.6 |
| ALT quotient ≤ 3.0, n (%) | 56 (75) | 52 (74) | 60 (83) | 63 (89) | 58 (83) | 58 (77) | 57 (79) |
| HCV subtype,* n (%) | |||||||
| 1a | 55 (74) | 45 (64) | 48 (67) | 47 (66) | 42 (61) | 45 (60) | 43 (60) |
| 1b | 18 (24) | 25 (36) | 21 (29) | 24 (34) | 26 (38) | 28 (37) | 29 (40) |
| Mean log10 HCV RNA ± SD, IU/mL | 6.3 ± 0.6 | 6.2 ± 0.6 | 6.3 ± 0.7 | 6.4 ± 0.6 | 6.4 ± 0.5 | 6.3 ± 0.7 | 6.3 ± 0.6 |
| HCV RNA ≥ 400,000 IU/mL, n (%) | 66 (89) | 62 (89) | 59 (82) | 61 (86) | 64 (91) | 65 (87) | 65 (90) |
| Fibrosis score,* n (%) | |||||||
| F0-2 | 55 (75) | 58 (84) | 51 (72) | 57 (81) | 56 (80) | 62 (86) | 59 (86) |
| F3-4 | 18 (25) | 11 (16) | 20 (28) | 13 (19) | 14 (20) | 10 (14) | 10 (14) |
The percentage of patients of African racial descent was higher in groups D (8%), F (9%), and G (7%) and lower (range 1 to 4%) in the other treatment groups, while the percentage of patients with advanced fibrosis was higher in groups A (25%) and C (28%) and lower (range 14 to 20%) in the other treatment groups (Table 1).
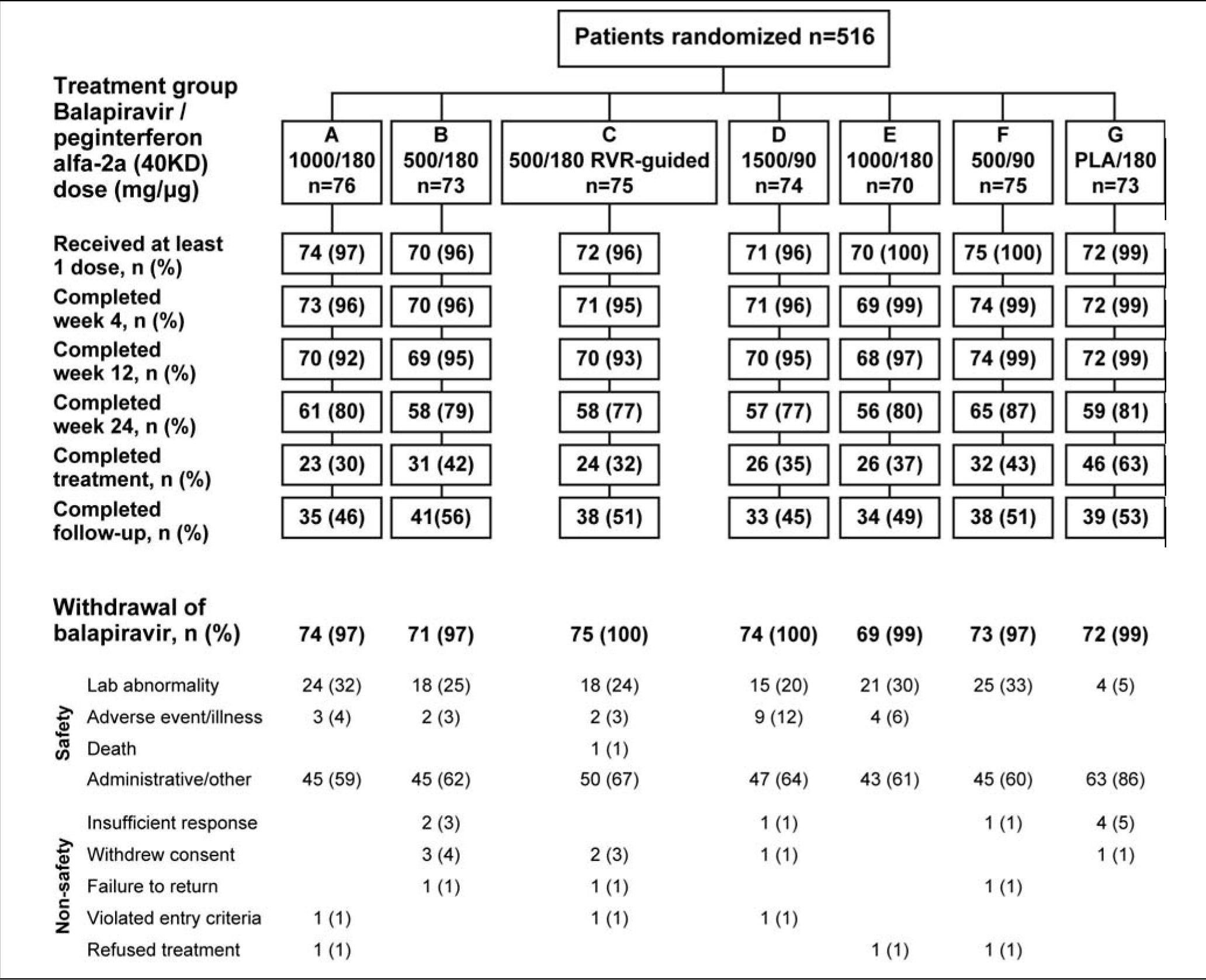
The flow of patients through the study is shown in figure 2. Following a recommendation from the data monitoring committee a protocol amendment resulted in termination of balapiravir treatment in patients who had received 12 or more weeks of treatment with the drug. Therefore, the most common reason for withdrawal from treatment with balapiravir is listed as administrative in figure 2.
, withdrawal of consent (n = 7) and administrative or other reasons (n = 2).")
Flow of patients through the trial showing the actual enrolment in each treatment group. Twelve patients were randomized but did not receive study medication because of violation of entry criteria (n = 3), withdrawal of consent (n = 7) and administrative or other reasons (n = 2).
Among patients randomized to placebo plus peginterferon alfa-2a (40KD) and ribavirin, mean reductions in serum HCV RNA concentration at week 2, 4, and 12 were 1.4 log10, 2.4 log10, and 3.7 log10, respectively. Treatment with balapiravir produced dose-dependent reductions in serum HCV RNA levels, with the highest mean reductions at week 2 (3.5 log10), week 4 (4.2 log10), and week 12 (4.9 log10) achieved in group A (balapiravir 1,000 mg bid plus peginterferon alfa-2a [40KD] 180 μg/ week in combination with ribavirin). At week 24 the reduction in HCV RNA level was similar in all treatment groups (range 4.0 log10 to 4.7 log10). HCV RNA concentration data were unavailable for a substantial proportion of patients (> 20%) after week 24.
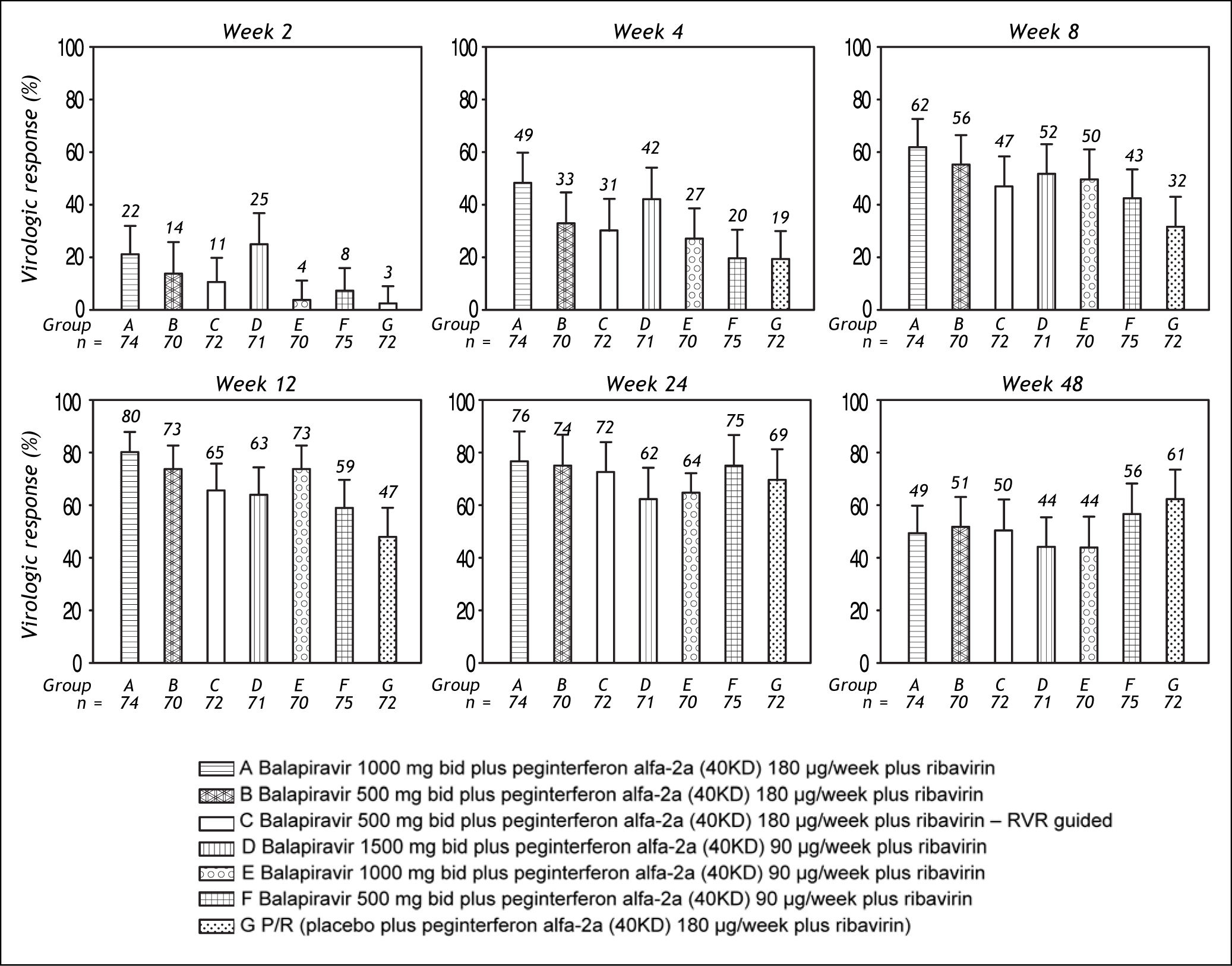
The percentage of patients with undetectable HCV RNA was consistently higher in all balapiravir treatment groups compared with the placebo plus peginterferon alfa-2a (40KD) and ribavirin group at treatment week 2, 4, 8, and 12; but was similar in all groups at treatment week 24 and 48 (Figure 3).
. Percentages are shown at the top of each bar. Vertical bars show the upper limit of the 95% confidence interval. Analyses were conducted according to the intention-to-treat principle.")
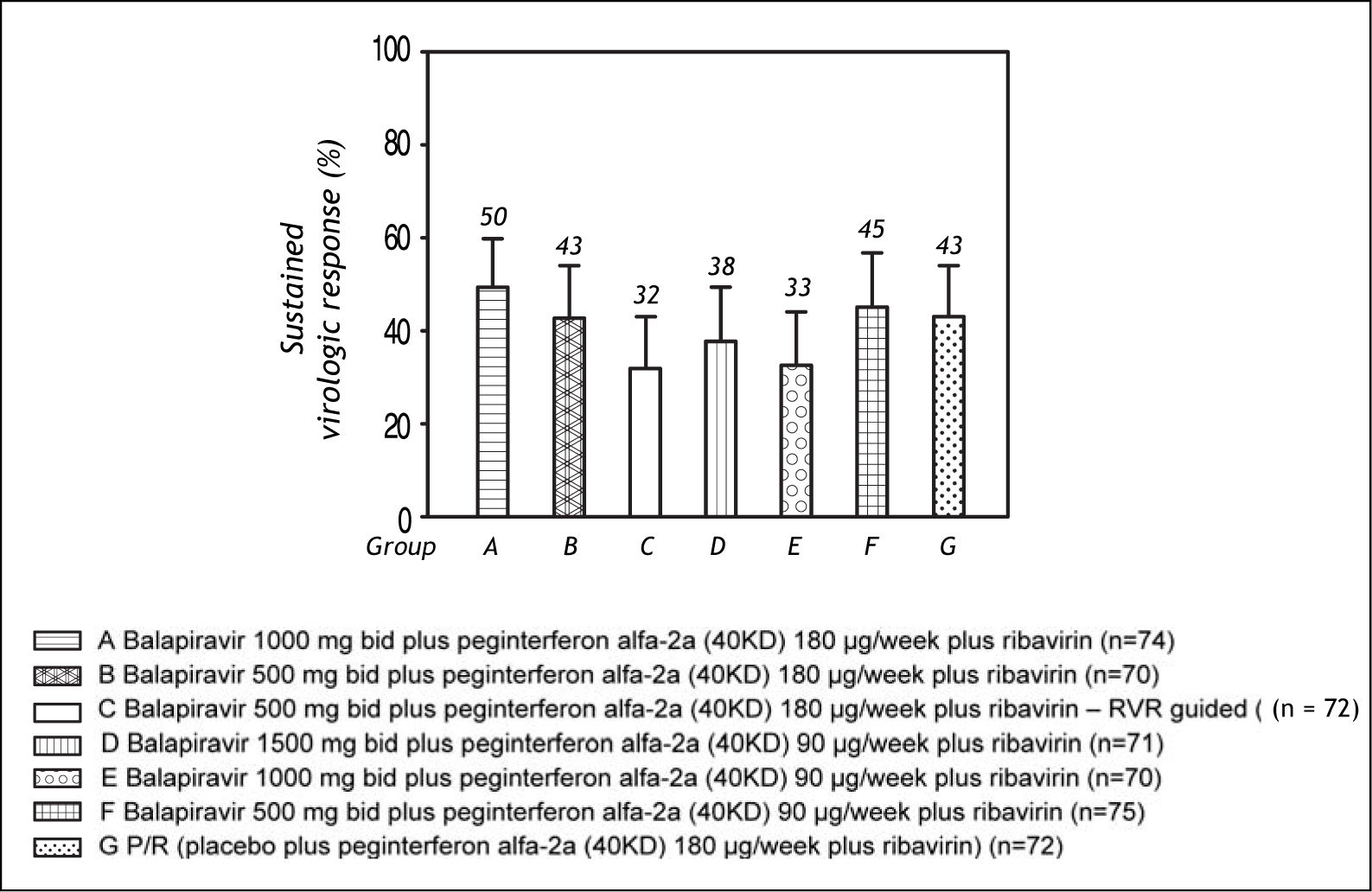
The SVR rate ranged from 32 to 50% across the balapiravir treatment groups and was 43% in the placebo plus peginterferon alfa-2a (40KD) and ribavirin group (Figure 4).
Safety in serum at the end of untreated follow-up. Vertical bars show the upper limit of the 95% confidence interval. Analyses were conducted according to the intention-to-treat principle.")
The percentage of patients withdrawn from treatment with study drug for safety reasons was higher in patients randomized to the balapiravir groups than in the placebo group. Among those randomized to groups A-F, treatment with balapiravir was withdrawn for safety reasons in 27 to 36% of patients, treatment with peginterferon alfa-2a (40KD) was withdrawn for safety reasons in 22 to 43% of patients, and treatment with ribavirin was withdrawn for safety reasons in 22 to 45% of patients. In contrast, among those randomized to group G (placebo plus peginterferon alfa-2a (40 KD) and ribavirin), placebo was withdrawn for safety reasons in 5% of patients, treatment with peginterferon alfa-2a (40KD) was withdrawn for safety reasons in 8% of patients, and treatment with ribavirin was withdrawn for safety reasons in 8% of patients.
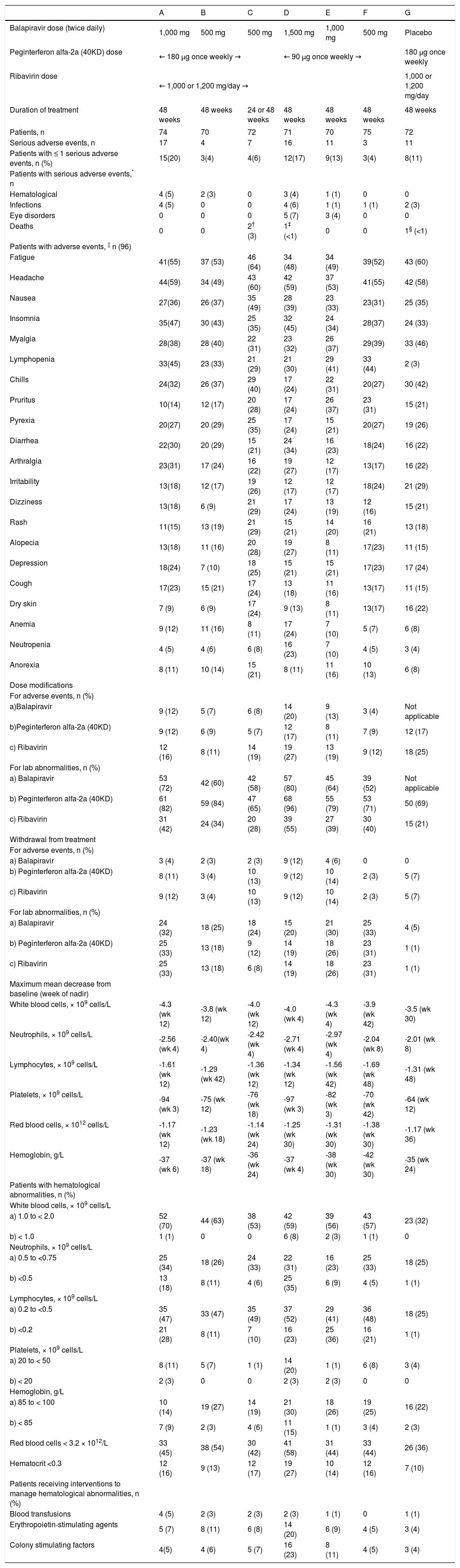
Among the balapiravir treatment groups, the number of serious adverse events and the percentage of patients with serious adverse events increased with the dose of balapiravir and was highest in groups A (balapiravir 1,000 mg bid plus peginterferon alfa-2a [40KD] 180 μg/week in combination with ribavirin), D (balapiravir 1,500 mg bid plus peginterferon alfa-2a [40KD] 90 μg/week in combination with ribavirin) and E (balapiravir 1,000 mg bid plus peginterferon alfa-2a [40KD] 90 μg/week in combination with ribavirin) (Table 2). Serious hematological adverse events and serious infections were more frequent in group A (5 and 5%, respectively) and group D (4 and 6% respectively) than in all other treatment groups in which the incidence of serious hematological adverse events and infections ranged from 0 to 3% (Table 2). Neutropenia and lymphopenia were observed during the study. Lymphopenia resolved in most patients after all study medication was stopped, but some patients remained lymphopenic more than 1 year after the end of treatment. Serious ocular adverse events were reported in five patients (7%) in group D and in three patients (4%) in group E (Table 2). The nature of these events was diverse and included optic neuropathy in two patients, and transient blindness, maculopathy, toxic optic neuropathy, reduced visual acuity, and optic neuritis in one patient each. All but one of these events was reported after more than 12 weeks of therapy (range: day 36 to 114 of treatment), all of these patients discontinued study medication, and six of eight events resolved during untreated follow-up. One patient has reported improvement and is still being followed (the remaining patient was lost to follow-up).
Adverse events, laboratory abnormalities and discontinuations. A B
| A | B | C | D | E | F | G | |
|---|---|---|---|---|---|---|---|
| Balapiravir dose (twice daily) | 1,000 mg | 500 mg | 500 mg | 1,500 mg | 1,000 mg | 500 mg | Placebo |
| Peginterferon alfa-2a (40KD) dose | ← 180 μg once weekly → | ← 90 μg once weekly → | 180 μg once weekly | ||||
| Ribavirin dose | ← 1,000 or 1,200 mg/day → | 1,000 or 1,200 mg/day | |||||
| Duration of treatment | 48 weeks | 48 weeks | 24 or 48 weeks | 48 weeks | 48 weeks | 48 weeks | 48 weeks |
| Patients, n | 74 | 70 | 72 | 71 | 70 | 75 | 72 |
| Serious adverse events, n | 17 | 4 | 7 | 16 | 11 | 3 | 11 |
| Patients with ≤ 1 serious adverse events, n (%) | 15(20) | 3(4) | 4(6) | 12(17) | 9(13) | 3(4) | 8(11) |
| Patients with serious adverse events,* n | |||||||
| Hematological | 4 (5) | 2 (3) | 0 | 3 (4) | 1 (1) | 0 | 0 |
| Infections | 4 (5) | 0 | 0 | 4 (6) | 1 (1) | 1 (1) | 2 (3) |
| Eye disorders | 0 | 0 | 0 | 5 (7) | 3 (4) | 0 | 0 |
| Deaths | 0 | 0 | 2† (3) | 1‡ (<1) | 0 | 0 | 1§ (<1) |
| Patients with adverse events, || n (96) | |||||||
| Fatigue | 41(55) | 37 (53) | 46 (64) | 34 (48) | 34 (49) | 39(52) | 43 (60) |
| Headache | 44(59) | 34 (49) | 43 (60) | 42 (59) | 37 (53) | 41(55) | 42 (58) |
| Nausea | 27(36) | 26 (37) | 35 (49) | 28 (39) | 23 (33) | 23(31) | 25 (35) |
| Insomnia | 35(47) | 30 (43) | 25 (35) | 32 (45) | 24 (34) | 28(37) | 24 (33) |
| Myalgia | 28(38) | 28 (40) | 22 (31) | 23 (32) | 26 (37) | 29(39) | 33 (46) |
| Lymphopenia | 33(45) | 23 (33) | 21 (29) | 21 (30) | 29 (41) | 33 (44) | 2 (3) |
| Chills | 24(32) | 26 (37) | 29 (40) | 17 (24) | 22 (31) | 20(27) | 30 (42) |
| Pruritus | 10(14) | 12 (17) | 20 (28) | 17 (24) | 26 (37) | 23 (31) | 15 (21) |
| Pyrexia | 20(27) | 20 (29) | 25 (35) | 17 (24) | 15 (21) | 20(27) | 19 (26) |
| Diarrhea | 22(30) | 20 (29) | 15 (21) | 24 (34) | 16 (23) | 18(24) | 16 (22) |
| Arthralgia | 23(31) | 17 (24) | 16 (22) | 19 (27) | 12 (17) | 13(17) | 16 (22) |
| Irritability | 13(18) | 12 (17) | 19 (26) | 12 (17) | 12 (17) | 18(24) | 21 (29) |
| Dizziness | 13(18) | 6 (9) | 21 (29) | 17 (24) | 13 (19) | 12 (16) | 15 (21) |
| Rash | 11(15) | 13 (19) | 21 (29) | 15 (21) | 14 (20) | 16 (21) | 13 (18) |
| Alopecia | 13(18) | 11 (16) | 20 (28) | 19 (27) | 8 (11) | 17(23) | 11 (15) |
| Depression | 18(24) | 7 (10) | 18 (25) | 15 (21) | 15 (21) | 17(23) | 17 (24) |
| Cough | 17(23) | 15 (21) | 17 (24) | 13 (18) | 11 (16) | 13(17) | 11 (15) |
| Dry skin | 7 (9) | 6 (9) | 17 (24) | 9 (13) | 8 (11) | 13(17) | 16 (22) |
| Anemia | 9 (12) | 11 (16) | 8 (11) | 17 (24) | 7 (10) | 5 (7) | 6 (8) |
| Neutropenia | 4 (5) | 4 (6) | 6 (8) | 16 (23) | 7 (10) | 4 (5) | 3 (4) |
| Anorexia | 8 (11) | 10 (14) | 15 (21) | 8 (11) | 11 (16) | 10 (13) | 6 (8) |
| Dose modifications | |||||||
| For adverse events, n (%) | |||||||
| a)Balapiravir | 9 (12) | 5 (7) | 6 (8) | 14 (20) | 9 (13) | 3 (4) | Not applicable |
| b)Peginterferon alfa-2a (40KD) | 9 (12) | 6 (9) | 5 (7) | 12 (17) | 8 (11) | 7 (9) | 12 (17) |
| c) Ribavirin | 12 (16) | 8 (11) | 14 (19) | 19 (27) | 13 (19) | 9 (12) | 18 (25) |
| For lab abnormalities, n (%) | |||||||
| a) Balapiravir | 53 (72) | 42 (60) | 42 (58) | 57 (80) | 45 (64) | 39 (52) | Not applicable |
| b) Peginterferon alfa-2a (40KD) | 61 (82) | 59 (84) | 47 (65) | 68 (96) | 55 (79) | 53 (71) | 50 (69) |
| c) Ribavirin | 31 (42) | 24 (34) | 20 (28) | 39 (55) | 27 (39) | 30 (40) | 15 (21) |
| Withdrawal from treatment | |||||||
| For adverse events, n (%) | |||||||
| a) Balapiravir | 3 (4) | 2 (3) | 2 (3) | 9 (12) | 4 (6) | 0 | 0 |
| b) Peginterferon alfa-2a (40KD) | 8 (11) | 3 (4) | 10 (13) | 9 (12) | 10 (14) | 2 (3) | 5 (7) |
| c) Ribavirin | 9 (12) | 3 (4) | 10 (13) | 9 (12) | 10 (14) | 2 (3) | 5 (7) |
| For lab abnormalities, n (%) | |||||||
| a) Balapiravir | 24 (32) | 18 (25) | 18 (24) | 15 (20) | 21 (30) | 25 (33) | 4 (5) |
| b) Peginterferon alfa-2a (40KD) | 25 (33) | 13 (18) | 9 (12) | 14 (19) | 18 (26) | 23 (31) | 1 (1) |
| c) Ribavirin | 25 (33) | 13 (18) | 6 (8) | 14 (19) | 18 (26) | 23 (31) | 1 (1) |
| Maximum mean decrease from baseline (week of nadir) | |||||||
| White blood cells, × 109 cells/L | -4.3 (wk 12) | -3.8 (wk 12) | -4.0 (wk 12) | -4.0 (wk 4) | -4.3 (wk 4) | -3.9 (wk 42) | -3.5 (wk 30) |
| Neutrophils, × 109 cells/L | -2.56 (wk 4) | -2.40(wk 4) | -2.42 (wk 4) | -2.71 (wk 4) | -2.97 (wk 4) | -2.04 (wk 8) | -2.01 (wk 8) |
| Lymphocytes, × 109 cells/L | -1.61 (wk 12) | -1.29 (wk 42) | -1.36 (wk 12) | -1.34 (wk 12) | -1.56 (wk 42) | -1.69 (wk 48) | -1.31 (wk 48) |
| Platelets, × 109 cells/L | -94 (wk 3) | -75 (wk 12) | -76 (wk 18) | -97 (wk 3) | -82 (wk 3) | -70 (wk 42) | -64 (wk 12) |
| Red blood cells, × 1012 cells/L | -1.17 (wk 12) | -1.23 (wk 18) | -1.14 (wk 24) | -1.25 (wk 30) | -1.31 (wk 30) | -1.38 (wk 30) | -1.17 (wk 36) |
| Hemoglobin, g/L | -37 (wk 6) | -37 (wk 18) | -36 (wk 24) | -37 (wk 4) | -38 (wk 30) | -42 (wk 30) | -35 (wk 24) |
| Patients with hematological abnormalities, n (%) | |||||||
| White blood cells, × 109 cells/L | |||||||
| a) 1.0 to < 2.0 | 52 (70) | 44 (63) | 38 (53) | 42 (59) | 39 (56) | 43 (57) | 23 (32) |
| b) < 1.0 | 1 (1) | 0 | 0 | 6 (8) | 2 (3) | 1 (1) | 0 |
| Neutrophils, × 109 cells/L | |||||||
| a) 0.5 to <0.75 | 25 (34) | 18 (26) | 24 (33) | 22 (31) | 16 (23) | 25 (33) | 18 (25) |
| b) <0.5 | 13 (18) | 8 (11) | 4 (6) | 25 (35) | 6 (9) | 4 (5) | 1 (1) |
| Lymphocytes, × 109 cells/L | |||||||
| a) 0.2 to <0.5 | 35 (47) | 33 (47) | 35 (49) | 37 (52) | 29 (41) | 36 (48) | 18 (25) |
| b) <0.2 | 21 (28) | 8 (11) | 7 (10) | 16 (23) | 25 (36) | 16 (21) | 1 (1) |
| Platelets, × 109 cells/L | |||||||
| a) 20 to < 50 | 8 (11) | 5 (7) | 1 (1) | 14 (20) | 1 (1) | 6 (8) | 3 (4) |
| b) < 20 | 2 (3) | 0 | 0 | 2 (3) | 2 (3) | 0 | 0 |
| Hemoglobin, g/L | |||||||
| a) 85 to < 100 | 10 (14) | 19 (27) | 14 (19) | 21 (30) | 18 (26) | 19 (25) | 16 (22) |
| b) < 85 | 7 (9) | 2 (3) | 4 (6) | 11 (15) | 1 (1) | 3 (4) | 2 (3) |
| Red blood cells < 3.2 × 1012/L | 33 (45) | 38 (54) | 30 (42) | 41 (58) | 31 (44) | 33 (44) | 26 (36) |
| Hematocrit <0.3 | 12 (16) | 9 (13) | 12 (17) | 19 (27) | 10 (14) | 12 (16) | 7 (10) |
| Patients receiving interventions to manage hematological abnormalities, n (%) | |||||||
| Blood transfusions | 4 (5) | 2 (3) | 2 (3) | 2 (3) | 1 (1) | 0 | 1 (1) |
| Erythropoietin-stimulating agents | 5 (7) | 8 (11) | 6 (8) | 14 (20) | 6 (9) | 4 (5) | 3 (4) |
| Colony stimulating factors | 4(5) | 4 (6) | 5 (7) | 16 (23) | 8 (11) | 4 (5) | 3 (4) |
One death was attributed to cardiac arrest unrelated to treatment in the opinion of the investigator in a patient with metastatic lymphoma in whom progression of lymphoma was considered to be possibly related to treatment; the second patient death was attributed to suicide (possibly related to treatment).
Four deaths were reported during treatment and follow-up. Three deaths were in balapiravir plus peginterferon alfa-2a (40KD) plus ribavirin combination treatment groups and were attributed to fulminant disseminated varicella possibly related to study medication (group D), cardiac arrest unrelated to study medication in a patient with lymphoma (group C), and suicide possibly related to study medication (group C). One death in the peginterferon alfa-2a (40KD) plus ribavirin control group was attributed to a road traffic accident unrelated to study medication (group G). Of note, the progression of disease in the patient with lymphoma in group C was classified as possibly related to study drug. It was considered likely that the lymphoma had been present but undetected prior to study start, based on the advanced stage of the cancer at diagnosis.
The most common adverse events were fatigue (range 48 to 64%), headache (49 to 60%), nausea (31 to 49%), insomnia (33 to 47%), and myalgia (31 to 46%), which occurred with similar frequency across the seven treatment groups (Table 2). Lymphopenia was a frequently reported adverse event in recipients of balapiravir (range 29 to 45%), but not in recipients of placebo (3%). Patients in group D also had considerably higher adverse event rates for anemia (24%) and neutropenia (23%) compared with all other treatment groups (Table 2).
Dose modifications of all three study drugs were more commonly attributed to laboratory abnormalities than adverse events in all treatment groups (Table 2). Balapiravir dose modifications for adverse events and laboratory abnormalities were most common in the highest dosage groups: A, D, and E. Peginterferon alfa-2a (40KD) and ribavirin dose modifications for laboratory abnormalities were also more common in these three treatment groups.
The maximum mean decrease from baseline in white blood cell count, lymphocyte count, neutrophil count, platelet count, red blood cell count, and hemoglobin concentration were generally greater in all balapiravir treatment groups than in the placebo plus peginterferon alfa-2a (40KD) and ribavirin group (Table 2).
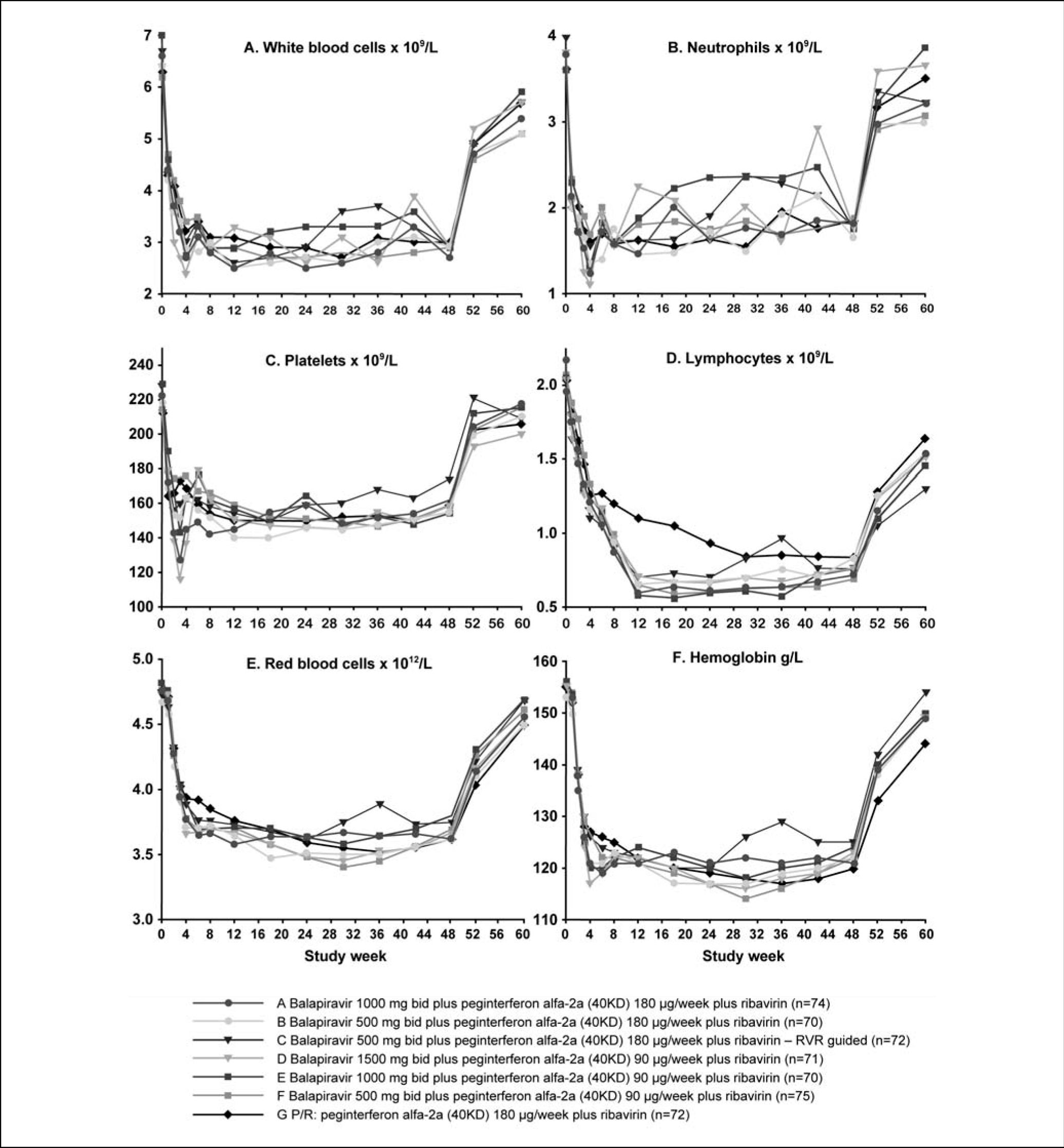
The time course of hematological parameters is shown in figure 5. The nadir in hematological parameters generally occurred earlier and was greater in all balapiravir treatment groups, with the exception of group F, than in the placebo plus peginterferon alfa-2a (40KD) and ribavirin group. The nadir in white blood cell count occurred between weeks 4 and 12 in groups A to E compared with week 30 with the placebo plus peginterferon alfa-2a (40KD) and ribavirin group, the nadir in neutrophil count occurred at week 4 in groups A to E and at week 18 with placebo plus peginterferon alfa-2a (40KD) and ribavirin, and the nadir in platelet count occurred at week 3 in groups A to E compared with week 12 with placebo plus peginterferon alfa-2a (40KD) and ribavirin. The lowest nadir for each of these parameters was observed in group D. Lymphocyte count declined markedly during the first 12 weeks of treatment and reached a nadir between weeks 12 and 24 in all balapiravir treatment groups. In contrast, the lymphocyte decline was less marked with placebo plus peginterferon alfa-2a (40KD) and ribavirin and did not reach a nadir until week 30. Lymphocyte count had not returned to baseline by week 60 in any group, including placebo plus peginterferon alfa-2a (40KD) and ribavirin. The red blood cell count and hemoglobin concentration declined rapidly during the first 4 weeks and then slowly thereafter to week 30 in all treatment groups with the exception of the RVR-guided treatment group C.

The highest incidence of specific laboratory abnormalities tended to occur in the highest balapiravir dose, i.e. group D (Table 2). The percentage of patients with grade 4 leucopenia (white blood cell count < 1.0 × 109/L), grade 3 or 4 lymphopenia (lymphocyte count < 0.5 × 109/L), grade 4 neutropenia (neutrophil count < 0.5 × 109/L), red blood cell count < 3.2 × 1012/L, and hematocrit < 0.3 were higher in all balapiravir treatment groups than in the placebo plus peginterferon alfa-2a (40KD) and ribavirin group. The percentage of patients with grade 4 thrombocytopenia (platelet count < 20 × 109/L) and a hemoglobin concentration < 85 g/L were higher in group A and D than in the placebo plus peginterferon alfa-2a (40KD) and ribavirin group. Of note, the incidence of grade 4 lymphopenia (lymphocyte count < 0.2 × 109/L) was higher in all balapiravir treatment groups (range 10 to 36%) than in the placebo plus peginterferon alfa-2a (40KD) and ribavirin group (1%).
DiscussionThe results of this trial demonstrate that the addition of balapiravir (RG1626) to peginterferon alfa-2a (40KD) plus ribavirin increases the rate and extent of the decrease in HCV RNA levels in patients with chronic hepatitis C genotype 1 infection. The decrease in HCV RNA levels was dose dependent, and a higher percentage of patients treated with balapiravir achieved an RVR at week 4 and a complete EVR at week 12 compared with peginterferon alfa-2a (40KD) plus ribavirin alone. However, the assessment of efficacy at later time points was hampered by the high rate of safety-related dose modifications and discontinuations that affected all three study drugs and was generally associated with temporary viral load rebounds during treatment. Subsequently, the higher rates of viral clearance observed during treatment were not maintained during untreated follow-up and there was no overall difference in SVR rates in the balapiravir combination treatment groups compared with placebo plus peginterferon alfa-2a (40KD) plus ribavirin.
The final SVR rate of 43% achieved with peginterferon alfa-2a (40KD) plus ribavirin alone in the trial is somewhat lower than that achieved with this combination in genotype 1 patients enrolled in phase 3 registration studies (46 to 52%),8,9 but falls within the range reported in other recent randomized studies in treatment-naive genotype 1 patients (41 to 55%).10-14
Treatment with balapiravir was not well tolerated and unexpected adverse events were observed in the present study. Hematological adverse events were anticipated and the protocol included several measures to mitigate against the incidence and severity of these events. The trial included two reduced dosage levels of peginterferon alfa-2a (40KD), and the highest dosage of balapiravir was administered with the lower dosage of peginterferon alfa-2a (40KD). Patients were monitored frequently and dose reduction protocols were provided for all study drugs. Despite these precautions a high proportion of patients treated with balapiravir developed anemia, neutropenia, lymphopenia, and thrombocytopenia, and study drug treatment was discontinued in a large number of these individuals because of laboratory abnormalities. In two of the highest balapiravir dosage groups (balapiravir 1,000 mg bid plus peginterferon alfa-2a [40KD] 180 μg/week and ribavirin; balapiravir 1,500 mg bid plus peginterferon alfa-2a [40KD] 90 μg/week and ribavirin), serious infections were reported in 5 to 6% of patients and one patient died from disseminated varicella infection.
The occurrence of late-onset serious ocular events in a small number of recipients of balapiravir was unexpected. The nature of these ocular events was diverse, and available data suggest that they are reversible. The mechanism that led to these events is under investigation.
Previous human studies of balapiravir include a dose escalation trial in which single doses as high as 12,000 mg were well tolerated in healthy volunteers,15 a 14-day trial of balapiravir monotherapy in patients with chronic hepatitis C at dosages of 500 mg to 4,500 mg bid,6 and a 28-day trial in which balapiravir was administered to patients with chronic hepatitis C at a dosage of 1,500 mg bid or 3,000 mg bid in combination with peginterferon alfa-2a (40KD) 180 μg/week or at a dosage of 1,500 mg bid in combination with peginterferon alfa-2a (40KD) plus ribavirin.7 In the 14-day monotherapy study no patients were withdrawn due to adverse events, and although dose-related hematological abnormalities were detected, they were described as being mild to moderate in severity.6 Hematological abnormalities were reported when balapiravir was administered in combination with peginterferon alfa-2a (40KD) with or without ribavirin in the 28-day study.7 The most common laboratory abnormalities in the trial were neutropenia and anemia, while lymphopenia was comparatively rare. Laboratory abnormalities were dose related and resolved after the end of treatment with balapiravir, and the rate of infections was similar in patients with mild or severe neutropenia.
On the basis of preclinical toxicity testing, the severity of hematological toxicity associated with longer-term administration of balapiravir in combination with peginterferon alfa-2a (40KD) and ribavirin in patients with chronic hepatitis C was unexpected. Balapiravir was well tolerated at doses up to 3,000 mg/kg/day (highest dose tested) for 3 months in mice, up to 2,000 mg/kg/day (highest dose tested) in rats for 6 months and up to 1,500 mg/kg/day (highest dose tested) for 9 months in female dogs. These studies had not identified any target organ toxicity.16 In vitro studies in human primary cells demonstrated measurable inhibition of bone marrow stem cell differentiation with low intrinsic cytotoxicity. Bone marrow stem cell differentiation into erythroid precursor cells was more sensitive to inhibition by balapiravir than was differentiation into myeloid or megakaryocytic lineages. Inhibition of erythroid differentiation by balapiravir was more than 80-fold less potent than that occurring with the comparator nucleoside analogue zidovudine.16 Consistent with the in vitro analysis, the most obvious dose-dependent hematological effect of balapiravir in patients treated with monotherapy was a reduction in hemoglobin concentration and erythrocyte counts that was reversible upon discontinuation, consistent with inhibition of precursor differentiation.6
The mechanism responsible for the hematological toxicity associated with balapiravir when administered in combination with peginterferon alfa-2a (40KD) and ribavirin is currently unknown, although it appears that this combination may have exacerbated a direct and broad-based suppressive effect of balapiravir on bone marrow and also resulted in an uncompensated reduction in lymphocytes. The hematological toxicity of balapiravir does not appear to be a class effect of HCV nucleoside polymerase inhibitors. Valopicitabine (NM283) was not associated with marked hematological adverse events when combined with peginterferon plus ribavirin; however, clinical development of this agent was halted because of severe gastrointestinal adverse events.17 Mericitabine (RG7128) is another nucleoside polymerase inhibitor currently being investigated in a phase 2b study to evaluate its safety and efficacy in triple combination therapy (500 mg or 1,000 mg bid with peginterferon alfa-2a [40KD] plus ribavirin) in HCV genotype 1 and 4 treatment-naive patients. A preliminary analysis of 12 weeks' safety data from 408 patients enrolled in this study has confirmed that mericitabine has a promising safety profile with no significant unexpected toxicities.18 To date no hematological, renal, gastrointestinal, dermatological, or other organ system events different than those expected with peginterferon alfa-2a (40KD) plus ribavirin have been detected.
Mericitabine has also been investigated in combination with an HCV protease inhibitor (danoprevir) in the proof of concept study of a dual interferonfree regimen for chronic hepatitis C (INFORM-1).19 The all oral direct acting antiviral regimen was effective and well tolerated in this study, in which 88 patients were treated for up to 13 days with mericitabine/danoprevir. No patient discontinued therapy because of adverse events, no treatment-related serious or severe adverse effects were reported and no grade 3 or 4 laboratory abnormalities were detected.19
Further development of balapiravir in combination with peginterferon alfa-2a (40KD) and ribavirin for the treatment of chronic hepatitis C has been halted because of the unacceptable safety profile revealed in this study. However, non-clinical studies are ongoing in an effort to identify the mechanism responsible for the hematological and ocular toxicities associated with the administration of balapiravir. Longer-term experience with other investigational nucleoside polymerase inhibitors is currently lacking and close monitoring of safety in ongoing studies is warranted.
Abbreviations- •
bid. Twice daily.
- •
HCV. Hepatitis C.
- •
RVR. Rapid virological response.
- •
SVR. Sustained virological response.
This work was supported in part by the NIH/ NCRR Clinical and Translational Science Award to the University of Florida UL1 RR029890.
The institutions and principal investigators who participated in this study are as follows:
Australia:- •
D. Crawford: Greenslopes Private Hospital, Greenslopes.
- •
J. George: Westmead Hospital, Westmead.
- •
S. Roberts: The Alfred Hospital, Melbourne.
- •
D. Shaw: Royal Adelaide Hospital, Adelaide.
- •
S. Strasser: Westmead Hospital, Westmead.
- •
P. Ferenci: AKH Wien Innere Medizin III, Wein.
- •
F. Anderson: Liver and Intestinal Research Centre, Vancouver.
- •
C. Cooper: The Ottawa Hospital, Ottawa.
- •
J. Heathcote: Toronto Western Hospital, Toronto.
- •
S. Lee: Heritage Medical Research Clinic, Calgary.
- •
M. Sherman: Toronto General Hospital, Toronto.
- •
E. Yoshida: The Gordon and Leslie Diamond Centre, Vancouver.
- •
M. Bourlière: Hôpital Saint-Joseph, Marseille.
- •
N. Boyer: Hôpital Beaujon, Clichy.
- •
J. Bronowicki: Hôpital de Brabois Adultes, Nancy.
- •
C. Hezode: Hôpital Henri Mondor, Créteil.
- •
V. Ledinghen: Hôpital Haut-Lévêque, Pessac.
- •
P. Marcellin: Hôpital Beaujon, Clichy.
S. Pol.: Hôpital Cochin, Paris.
- •
T. Berg: Universitätsmedizin Berlin, Berlin.
- •
P. Buggisch: I. Medizinische Klinik und Poliklinik, Hamburg.
- •
A. Lohse: I. Medizinische Klinik und Poliklinik, Hamburg.
- •
T. Goeser: Klinikum der Universität zu Köln, Köln.
- •
M. Manns: Medizinische Hochschule, Hannover.
- •
J. Rasenack: Medizinische Universitätsklinik Freiburg, Freiburg.
- •
S. Zeuzem: Klinikum der Johann Wolfgang Goethe Universität, Frankfurt.
- •
P. Andreone: Dipartimento Malattie Apparato Digerente e Medicina Interna, Bologna.
- •
A. Ascione: Struttura Complessa di Gastroenterologia ed Epatologia, Milano.
- •
F. Lampasi: Struttura Complessa di Gastroenterologia ed Epatologia, Milano.
- •
M. Rizzetto: Università di Torino, Torino.
- •
M. Diago-Madrid: Hospital General Universitario de Valencia, Valencia.
- •
J. Luis-Calleja: Hospital Puerta de Hierro, Madrid.
- •
R, Planas-Vila: Hospital Universitario Germans Trias i Pujol, Barcelona.
- •
M. Romero-Gómez: Hospital Nuestra Señora de Valme, Seville.
- •
R. Sola-Lamoglia: Hospital del Mar, Barcelona.
- •
D. Bernstein: North Shore-Long Island Health System, Manhasset.
- •
T. Box: Mountain West Gastroenterology, Utah.
- •
N. Bräu: Bronx VA Medical Center, New York.
- •
N. Bzowej: California Pacific Medical Center, California.
- •
G. Everson: University of Colorado Denver, Colorado.
- •
M. Fried: Univ. of North Carolina at Chapel Hill, North Carolina.
- •
R. Ghalib: The Liver Institute at Methodist Hospital, Texas.
- •
E. Godofsky: University Hepatitis Center at Bach & Godofsky, Florida.
- •
S. Harrison: Brooke Army Medical Center, Texas.
- •
T. Hassanein: University of California San Diego Medical Center, San Diego.
- •
R. Herring: Nashville Gastrointestinal Specialists, Inc., Nashville.
- •
I. Jacobson: Weill Medical College of Cornell University, New York.
- •
D. Jensen: University of Chicago, Chicago.
- •
M. Kugelmas: South Denver Gastroenterology, Denver.
- •
E. Lawitz: Alamo Medical Research, San Antonio.
- •
T. Morgan: VA Long Beach Healthcare System, Long Beach.
- •
D. Nelson: University of Florida Hepatology, Florida.
- •
L. Nyberg: Kaiser Permanente, San Diego.
- •
C. O'Brien: University of Miami, Miami.
- •
P. Pockros: Scripps Clinic, San Diego.
- •
M. Rodríguez-Torres: Fundación de Investigación de Diego, San Juan.
- •
L. Rossaro: University of California, Sacramento.
- •
V. Rustgi: Metropolitan Research, Fairfax.
- •
T. Sepe: University Gastroenterology, Rhode Island.
- •
M. Shiffman: McGuire DVAMC, Virginia.
- •
K. Sherman: University of Cincinnati College of Medicine, Cincinnati.
- •
C. Smith: Minesota Gastroenterology, Minesota.
- •
J. Smith: Penn State Milton S. Hershey Medical Center, Hershey.
- •
M. Sulkowski: Johns Hopkins University, Maryland.
- •
A. Sunyal: McGuire DVAMC, Virginia.
- •
I. Thomason: Mountain West Gastroenterology, Utah.
- •
H. Vargas: Mayo Clinic Arizona, Arizona.
- •
A. Zaman: Oregan Health and Science University, Oregan.
Grant Support: This study was funded by Roche. The sponsor contributed to study design as well as data collection, analysis, and interpretation with input from the investigators. Writing support was provided by Blair Jarvis of Health Interactions and funded by Hoffman La-Roche Ltd. ClinicalTrials.gov Identifier: NCT00517439



