



Hyperammonemia and associated cerebral edema cause neurological abnormalities in liver disease patients. Although only 15% of ammonia production originates in the colon, management strategies for hepatic encephalopathy (HE) have focused on reducing ammonia generation from the bowel rather than on manipulating systemic mechanisms involved in ammonia metabolism. Administration of L-ornithine L-aspartate (LOLA) improves mental status and decreases serum and spinal fluid ammonia levels by stimulating both the urea cycle and glutamine (Gln) synthesis, which are key metabolic pathways in ammonia detoxification. LOLA was shown to be superior to a placebo for management of HE, and the results of several clinical trials suggest that its effectiveness could be higher with the more severe grades of this syndrome. Compared with the standard treatment, LOLA is effective not only in reducing hyperammonemia and the severity of this disease, but also in improving the patient's perceived quality of life. Therefore, LOLA is a promising alternative for the management of HE.
Although the association between neurological abnormalities and liver disease was recognized by Hippocrates in the 5th century B.C., the pathophysiological mechanism responsible for these abnormalities remained obscure until the late 19th century. Liver dysfunction limits the metabolism of substances that are neurotoxic at high concentrations. The neurological manifestations of liver disease range from subtle changes in mental status to coma. This set of neurological changes is called HE.
HE and its association with decompensated liver disease is important because it has a negative impact on patient prognosis and quality of life, and treatment is costly. This syndrome affects up to 50% of cirrhotic patients with advanced disease.1 After the first episode of HE, the 1-year survival rate is 42% and the 3-year survival rate is 23%.2 In 2003, the total hospital cost associated with HE in the United States was estimated to be $900 million.3
Ammonia is the neurotoxin that triggers HE syndrome. To date, treatment modalities have focused on reducing ammonia produced by the bowel rather than on manipulating mechanisms involved in the systemic production of ammonia.
Despite the limited number of clinical trials with high internal validity that support the use of nonabsorbable disaccharides (NADs) for treating HE, NADs are still considered the drugs of choice.4 LOLA decreases ammonia concentration by stimulating the urea cycle and expression of glutamine synthetase (GS). In cirrhotic patients, the synthesis of Gln via GS represents an alternative pathway for detoxification of ammonia.
Pathogenesis of Hepatic EncephalopathyFor many years, it was thought that most of the ammonia in the body was produced by degradation of nitrogenous products by the colonic flora. However, current evidence indicates that 85% of the intestinal production of ammonia is the result of phosphate-activated glutaminase activity in the small intestine.5,6 The kidney also plays an important role in the generation of ammonia, and may contribute one-third or more of the ammonia released into the splanchnic circulation.7
In healthy subjects, the main mechanisms involved in maintaining blood ammonia concentration within nontoxic limits are the urea production via the Krebs cycle and GS-mediated synthesis of Gln in the liver.8 In cirrhotic patients, a reduction in the hepatocellular function and the spontaneous generation of portosystemic shunts are responsible for hyperammonemia.9 In this situation, the processing of ammonia and its conversion into a nontoxic compound are carried out through Gln synthesis in the liver, brain and muscles.8,10
Ammonia passes freely through the blood–brain barrier (BBB), and is required for the production of Gln in astrocytes. The uptake of glutamate by the astrocyte and the production of Gln from glutamate and ammonia prevent excessive neuronal activation in healthy subjects.11,12 In liver disease, neurological abnormalities are associated with low-grade cerebral edema, which is secondary to hyperammonemia. The brain and spinal fluid of cirrhotic patients with hyperammonemia contain excessively high levels of Gln. Accumulation of ammonia and Gln in astrocytes results in oxidative stress, free radical formation and mitochondrial and sodium channel dysfunction, which ultimately increase intracellular osmolarity, causing edema and cerebral malfunction.13-15 Consequently, brain GS does not contribute to the detoxification of ammonia. However, the contribution of muscle GS to ammonia detoxification is substantial because this tissue constitutes a significant proportion of total body mass, and the Gln so produced is the major substrate for the generation of ammonia in the kidney.10,16 Hyperammonemia reduces the release of ammonia from the kidneys to the splanchnic circulation and increases urinary excretion of ammonia by up to 70%; that is, it becomes an ammonia-excreting organ and, therefore, is responsible for the systemic removal of this neurotoxin.10,17
Mechanism of Action of L-Ornithine L-AspartateClinical studies supporting the use of LOLA in humans for treating HE began in Germany almost 40 years ago. LOLA is a salt of the natural amino acids ornithine and aspartic acid, and provides key substrates to metabolic pathways involved in the detoxification of ammonia.18,19 The administration of LOLA improves mental status and decreases ammonia levels in serum and spinal fluid by stimulating the urea cycle and the synthesis of Gln. After administration of LOLA, normalization of plasma ammonia levels is concomitant with a decrease in brain water content, which delays the onset of neurological symptoms.20
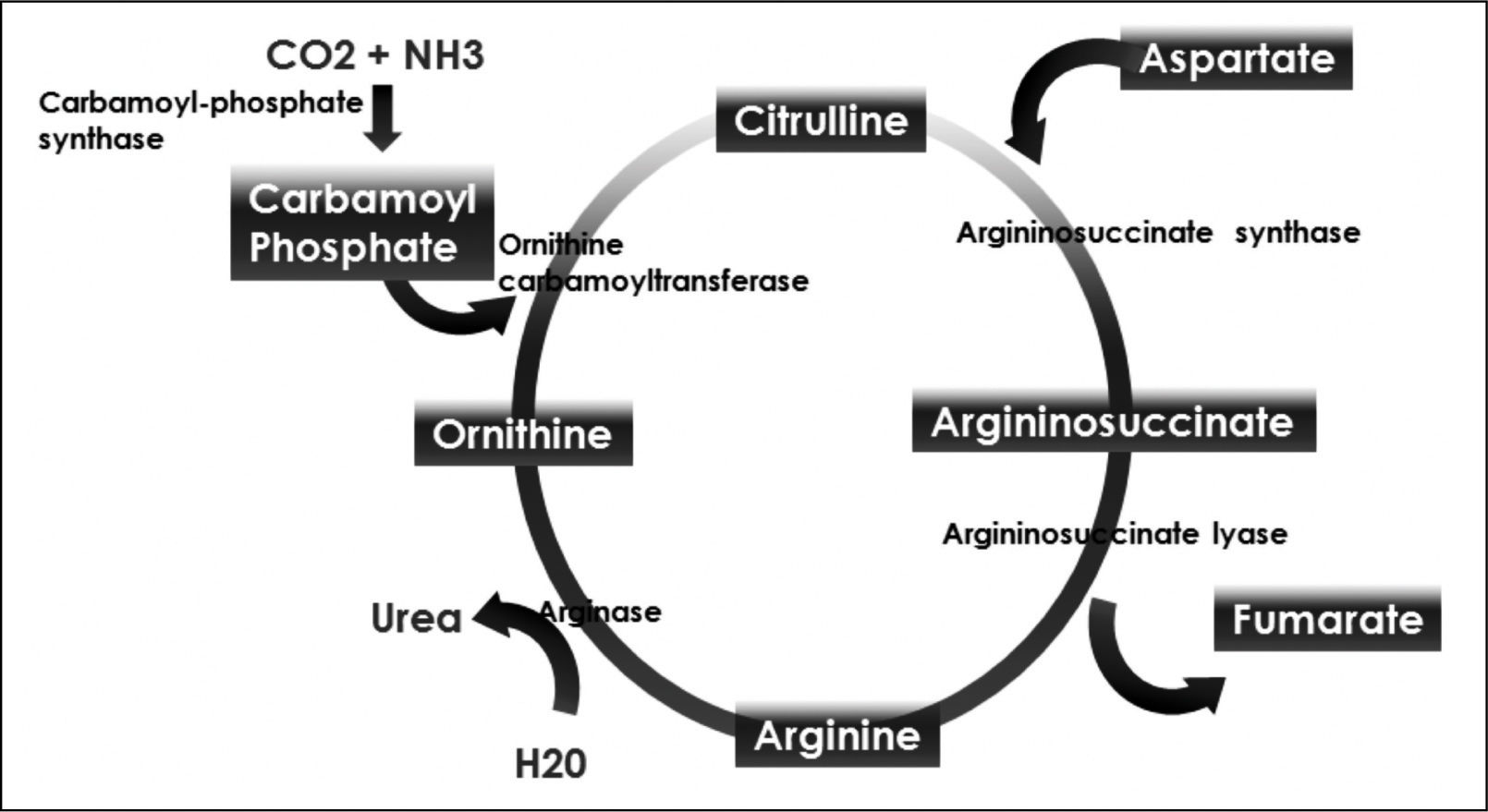
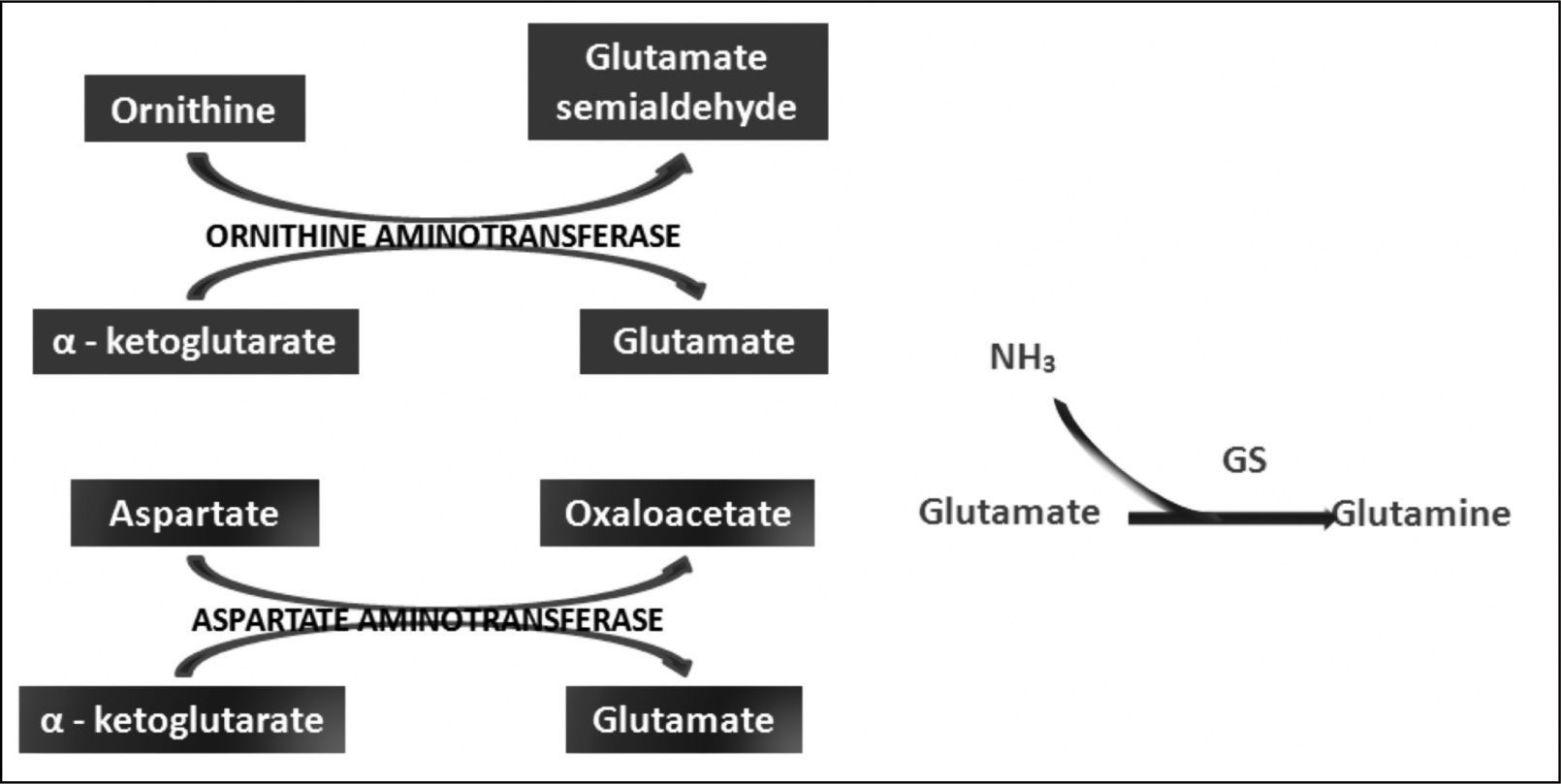
Ornithine stimulates the activity of carbamoyl phosphate synthetase I and aspartate stimulates the activity of arginase by donating nitrogen (Figure 1). Both of these enzymes are necessary for the synthesis of urea. The administration of LOLA decreases the plasma concentration of ammonia and increases the plasma concentration of urea, which proves that LOLA increases the activity of the Krebs cycle.21,22 When liver function is impaired, ammonia that cannot be metabolized by the liver is converted into Gln in the muscle. Thus, Gln functions as a nontoxic ammonia transporter in the circulation.7,9,10 After administration of LOLA, serum Gln levels increase because of the activity of muscle GS. However, levels of Gln and lactate in spinal fluid are not increased, which prevents the onset of cerebral edema. This supports that LOLA increases Gln synthesis in the periphery.20 The primary mechanism of detoxification of ammonia in cirrhotic patients is the uptake of ammonia that escapes the liver by muscle and its subsequent conversion to Gln in muscle.7,16 LOLA enhances the action of ornithine and aspartate transaminases to produce glutamate, which then promotes the synthesis of Gln by GS21 (Figure 2).


Ornithine passes through the BBB, suggesting that the central nervous system is a target for ornithine, but the mechanism by which it exerts effects is unknown. The improvement in mental status after therapy with LOLA is the result not of a direct effect of ornithine on the central nervous system, but of a decrease in exposure of the brain to ammonia secondary to a decrease in serum ammonia level.22
EfficacyAs HE is diagnosed clinically, mental status is considered the primary outcome measure for establishing treatment efficacy in clinical trials. Objective clinical evaluation of HE is accomplished by analyzing the dominant frequency of an electroencephalogram, P300 auditory evoked potentials and the portosystemic encephalopathy index (PSEI). This index comprises electroencephalogram results, number connection test (NCT) results, the degree of asterixis, serum ammonia levels and the results of mental status evaluation. As minimal hepatic encephalopathy (MHE) has no apparent manifestations to clinical observers, the primary outcome measure is based on the results of neuropsychological and neurophysiological tests.23
HE is in some cases episodic and resolved by removing the triggering event, making it essential that a drug show efficacy against a placebo before its effects are compared with those of an active control. The superiority of LOLA over placebo as an oral treatment for the management of HE has been demonstrated in human models.24-26
The bioavailability of LOLA when administered p.o. is 82.2 ± 28%,27 and the efficacy of its administration by this route was supported by Stauch, et al.25 This group of researchers compared the effectiveness of p.o. LOLA administered with a placebo in preventing hyperammonemia induced by a high-protein diet in 66 patients with chronic grade I or grade II HE or MHE according to the West Haven Criteria. Eighteen grams of LOLA or 10 g of fructose was given daily for 14 consecutive days. The primary outcome measures were postprandial ammonia concentration and NCT performance, and the secondary outcome measures were mental status and PSEI. LOLA was superior to the placebo in reducing postprandial ammonia level (p < 0.05) and in improving NCT performance (p < 0.01), mental status (p < 0.05) and PSEI (p < 0.01). Subgroup analysis showed that the most pronounced clinical response to LOLA occurred in patients with grade II HE (57% of patients in LOLA group, whereas only 18% in the placebo group responded). In contrast, only 9% of patients with MHE showed a response to LOLA, suggesting that the more severe the impairment of mental status, the greater is the effect of LOLA. It is likely that the lack of response in the MHE group was a function of the method used for diagnosis of this condition. The use of a set of standardized neuropsychological tests or neuropsychological tests in conjunction with P300 or electroencephalogram dominant frequency tests, which are capable of detecting subtle neurological changes in the early stages of the disease, is recommended.
Kircheis, et al.19 compared the effect of intravenous LOLA with that of a placebo in 126 cirrhotic patients with hyperammonemia and MHE or grade I or II HE according to the West Haven Criteria. LOLA was superior to the placebo with respect to NCT performance (p < 0.001), postprandial ammonia level (p < 0.001), preprandial ammonia level (p < 0.01), mental status (p < 0.001) and PSEI (p < 0.01). The differences were more pronounced in patients with grade I or grade II HE than in patients with MHE, and, unlike Stauch, et al., they used both NCT and electroencephalography to diagnose MHE. This confirms the finding by Stauch that the greater the impairment in mental status, the greater the impact of LOLA.
NADs are considered the drugs of first choice for treating HE. NADs act on the colon, shortening intestinal transit time and decreasing colon pH, which reduces the absorption of nonionized ammonia and increases the assimilation of ammonia by bacteria.28 As the evidence shows that only 15% of ammonia production originates from the colon, the contribution of NADs to the reduction of hyperammonemia is limited.
To compare the efficacy of oral LOLA with that of lactulose, 20 patients with grade I or grade II HE were randomized by Po, et al., to receive 30 mL of lactulose or 9 mg of LOLA orally for 2 weeks. The doses could be adjusted up to 60 mL of lactulose and up to 18 mg of LOLA, according to researcher’s opinion.24 The baseline ammonia concentration decreased from 120.4 ± 8.1 to 91.4 ± 10 μg/dL (p < 0.05) in the lactulose group, and from 141.6 ± 9.1 to 96.9 ± 9.3 μg/dL (p < 0.05) in the LOLA group. Although lactulose decreased baseline ammonia concentration, it did not affect the other variables used in the PSEI. In contrast, after administration of LOLA, a significant improvement in mental status, NCT, asterixis, and in the electroencephalogram activity, was observed (p < 0.05). The difference between PSEI values at baseline and those measured after 2 weeks in the LOLA group reach statistical significance (0.44 ± 0.03 and 0.28 ± 0.04, respectively; p < 0.05). Improvement in the quality of life was assessed using a EuroQol visual analog scale. The EuroQol index improved after both interventions (p < 0.05), the baseline and final indexes were 51.1 ± 24.1 and 61.5 ± 15.8, for the lactulose group and 56.5 ± 24.5 and 70 ± 19.4, for the LOLA group.
A meta-analysis designed to determine the efficacy and safety of LOLA for management of HE29 included the studies of Kircheis, Stauch, and Poo and analyzed data from 212 patients.19,24,25 According to this report, compared with the placebo, LOLA resulted in clinical improvement of HE (RR, 1.89; 95% CI, 1.32-2.71; p = 0.0005). Subgroup analysis showed that LOLA was superior to the placebo in grade I and grade II HE (RR = 1.87; 95% CI, 1.30-2.68; p < 0.0007), but had no significant effect in patients with MHE (RR = 1.69; 95% CI, 0.72-3.94; p = 0.23).
ConclusionsCurrently, HE therapy is focused solely on reducing the amount of ammonia produced in the colon. However, evidence suggests that only 15% of ammonia of intestinal origin is produced in the colon. Therefore, there is no valid basis for using NADs as monotherapy for HE. LOLA promotes activation of the main ammonia detoxification routes and ammonia storage in the muscles as glutamine, a nontoxic carrier compound. Therefore, LOLA is involved in the systemic elimination of ammonia, making it an excellent therapeutic alternative.
The bioavailability of p.o. LOLA is 82.2 ± 28%,27 enabling it to be administered by this route without sacrificing efficacy. There is enough evidence that supports the superiority of p.o. LOLA over placebo for the management of HE. The studies of Stauch and Kircheis suggest that LOLA efficacy is greater in the more severe forms of the syndrome and warrants further examination of the effects of LOLA in grades III and IV of HE.
Controlled clinical trials comparing p.o. LOLA with standard therapy show that LOLA is as effective as NADs in the management of HE. The main impact of HE is not the treatment cost, but decreased survival and quality of life. The administration of LOLA has been proven effective, not only in reducing hyperammonemia and the severity of this disease, but also in improving the patient's perceived quality of life.
Abbreviations- •
HE: Hepatic encephalopathy.
- •
LOLA: L-ornithine L-aspartate.
- •
NADs: Nonabsorbable disaccharides.
- •
GS: Glutamine synthetase.
- •
Gln: Glutamine.
- •
PSEI: Portosystemic encephalopathy index.
- •
NCT: Number connection test.
- •
MHE: Minimal hepatic encephalopathy.



