



The occurrence of hepatocellular carcinoma (HCC) is not entirely clear at present. This study comprehensively described the landscape of genetic aberrations in Chinese HCC patients using next-generation sequencing (NGS) and investigated the association of genetic aberrations with clinicopathological characteristics and prognosis.
Materials and MethodsThe clinicopathological data of 78 HCC patients undergoing surgery were retrospectively analyzed. The genomic DNA extracted from tumor samples was detected using a NGS-based gene panel.
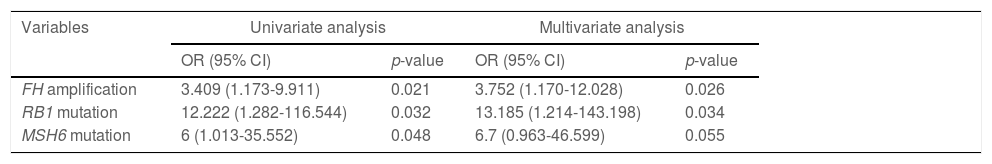
ResultsMutations in TP53 (55%), TERT (37%), MUC16 (29%) and CTNNB1 (27%) were most common in HCC. The co-occurrences between frequently mutated genes occurring ≥10% were relatively common in HCC. Forty-eight (61.5%) cases harbored DNA damage repair gene mutations, mainly including PRKDC (11.5%), SLX4 (9.0%), ATM (7.7%), MSH6 (7.7%), and PTEN (6.4%), and 39 (50.0%) patients had at least one actionable mutation. FH amplification (odds ratio: 3.752, 95% confidence interval: 1.170-12.028, p=0.026) and RB1 mutations (odds ratio: 13.185, 95% confidence interval: 1.214-143.198, p=0.034) were identified as the independent risk factors for early postoperative recurrence in HCC.
ConclusionsOur study provides a novel insight into the genomic profiling of Chinese HCC patients. FH amplification and RB1 mutations may be associated with an increased risk of early postoperative recurrence in HCC.
Hepatocellular carcinoma (HCC), a highly heterogeneous malignancy, is the second and the sixth leading cause of cancer deaths in males and females worldwide, respectively [1]. Its occurrence is associated with a variety of factors, including chronic hepatitis B (HBV) and C (HCV) infection, alcohol abuse, fatty liver disease and other genetic disorders [2]. Despite great advances in HCC management, most patients are diagnosed at advanced stages with limited therapeutic options. At present, several treatment regimens, including tyrosine kinase inhibitors (sorafenib, lenvatinib, regorafenib, and cabozantinib), monoclonal antibodies against vascular endothelial growth factor receptors (VEGFR2 and ramucirumab) and immune checkpoint inhibitors (nivolumab, pembrolizumab) are used to improve the survival in advanced HCC, but they are found to have a series of adverse reactions, limited survival benefits and primary or secondary resistance [3–5]. For developing more potent drugs to improve survival, more evidence on the HCC pathogenesis is still needed.
The genomic landscape of HCC has been described precisely with the development of next-generation sequencing (NGS). Accumulation of somatic genomic aberrations in regulatory pathways plays an important role in the carcinogenic process [2]. For each tumor, most mutations occur in passenger genes, whereas only 2-6 are the driver mutations with significant effects on the tumor evolution [2,4]. In HCC, TERT (promotor), CTNNB1, TP53, AXIN1, ARID2 and ARID1A genes are the most mutated driver genes, while others account for less than 10% of mutations [4]. These genes can be mainly categorized into several biological pathways, including telomere maintenance, cell cycle regulation, Wnt/β-catenin, epigenetic modifiers, mitogen-activated protein kinase, v-akt murine thymoma viral oncogene homolog /mechanistic target of rapamycin and oxidative stress [6–10].
A previous study showed that whole-genome sequencing analysis of HCC contributed to identifying etiological impacts on the patterns of somatic mutations and recurrent mutations in chromatin regulators [8]. Through whole exome sequencing and DNA copy number analysis, LZTR1, EEF1A1, SF3B1, and SMARCA4 were found to be significantly mutated in HCC, and one subtype based on the integrative molecular subtyping was associated with poorer prognosis [11]. Additionally, a hybridization capture-based NGS assay was also confirmed to be capable of providing predictive and prognostic genetic information for contemporary therapies of HCC patients and identifying potentially actionable mutations [12]. However, there is a lack of studies on the genomic landscape of the Chinese HCC population. Therefore, we attempted to describe the landscape of genetic aberrations in Chinese HCC patients using NGS, and analyzed the association of genetic aberrations with clinicopathological characteristics and prognosis, with the purpose of accelerating the identification of potential predictive biomarkers of response to immunotherapy and targeted therapy.
2Materials and Methods2.1Patients and tissue samplesA series of 90 primary HCC tissue samples were collected from the patients surgically treated in Shanghai Eastern Hepatobiliary Hospital between June 2019 and December 2020. All the formalin-fixed paraffin-embedded (FFPE) tumor samples stored at room temperature were processed within three days of collection.
2.2Data collectionThe clinicopathological data of patients were extracted by reviewing the electronic medical record, including gender, age, tumor size, Barcelona Clinic Liver Cancer (BCLC) staging, TNM staging, microvascular invasion (MVI), presence or absence of HBV/HCV infection, liver cirrhosis and portal vein tumor thrombus (PVTT), as well as alpha-fetoprotein (AFP), carcinoma embryonic antigen (CEA), carbohydrate antigen 19-9 (CA19-9) and protein induced by vitamin K absence/antagonist-II (PIVKA-II) levels.
2.3Panel-based sequencingRoutine immunohistochemical staining was performed. The antibodies for programmed cell death ligand-1 (PD-L1) were 22C3 (product number: M3653), which was provided by DAKO Company. At least two gastrointestinal pathologists were responsible for reviewing and confirming the tumor samples. If there were different opinions, a gastrointestinal pathology consensus conference would be held. FFPE tissue blocks close to the center of the tumor were sectioned at 5-20 μm and macrodissected in due time after pathologic review. DNeasy Blood & Tissue Kits from Qiagen were used for tissue deparaffinating and DNA extraction. DNA concentration was detected by a Qubit Fluorometer and the Qubit ds DNA HS (high sensitivity) Assay Kit, which was provided by Invitrogen Corporation, USA.
NGS was used to analyze the genomic DNA from tumor samples. The standard input of DNA was 50 ng, with the minimum input of 20 ng in some cases where DNA quantity was limited. Through custom oligonucleotide probes, barcoded DNA libraries from tumor and normal samples were captured, sequenced on an Illumina NovaSeq 6000 apparatus using Illumina 2 × 150-bp paired-end reads, and subjected to a custom analysis pipeline to identity somatic mutations like copy number variations (CNVs), small insertions and deletions (InDels) and single nucleotide variants (SNVs), and selected structural rearrangements in all exons and selective introns of 539 cancer-related genes [13]. Detection of somatic mutations was performed with the lowest sequencing depth of 100X and variant allele frequency of 1%. For the detection of CNVs, the amplification within the whole genome was analyzed, with three copies as a value of gene amplification. The fragments with CNVs were detected based on on-target and off-target reads, and the results were output after screening and filtration. The somatic SNVs and small InDels were identified using Vardict (version 1.5.7).
2.4Assessment of tissue tumor mutation burden (tTMB) and PD-L1 expressionThe tTMB was computed on 1Mb of genomic coding region through a panel including 539 cancer-related genes. The value of ≥10 mutations/Megabase (mut/Mb) was defined as the high tTMB (tTMB-high), while that of <10 mut/Mb was as the low tTMB (tTMB-low) [14]. The percentage of PD-L1 positive tumor cells (TC value) was calculated using an immunohistochemistry assay, and PD-L1 positivity was defined at the time of TC value ≥1 [15].
2.5Filtration of genomic aberrations using OncoKBOncoKB, a precision oncology knowledge base that can provide evidence-based information for somatic mutations, was used to filter genomic aberrations for oncogenic variants [16]. At different therapeutic levels of OncoKB (version 3.5), the number of genes was different, including 42 genes at level 1, 17 genes at level 2, 25 genes at level 3 and 23 genes at level 4, and these genes were all included in our panel. The level of evidence scales 1-4 for genomic aberrations was considered as actionable, in which level 1-2 represented a standard therapeutic intervention, level 3A-3B indicated investigational therapeutic options, and level 4 showed hypothetical therapeutic alterations.
2.6Statistical analysisThe data were analyzed using R package (version 4.0.2) and GraphPad Prism (version 8.0.1; GraphPad Software, San Diego, CA, USA). The normality test was used to judge whether the data conformed to normal distribution. The normally distributed measurement data were compared by t-test, presenting as the mean±standard deviation (x¯±s), while those with abnormal distribution were compared using Mann-Whitney U rank sum test, manifesting as the median and interquartile [M (Q1, Q3)]. Enumeration data compared by Chi-square test or Fisher's exact test were shown as n(%). Correlation between molecular data and clinicopathological features was analyzed using Chi-square test. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment was analyzed using a hypergeometric test and Bonferroni correction. The variables with a significant difference in univariate analysis were enrolled into a multivariate Logistic regression model to investigate the predictors for early postoperative recurrence. The two-sided p-value <0.05 represented statistically significant.
2.7Ethical statementWritten informed consent was obtained from each patient. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of Shanghai Eastern Hepatobiliary Hospital (approval number: EHBHKY2021-K-008).
3Results3.1Description of patientsBetween June 2019 and December 2020, a total of 90 HCC patients underwent surgery. After excluding 3 cases of stage III-IV, 2 cases of recurrence prior to surgery, 1 case of non-early recurrence (recurrence two years after surgery), 4 cases of incomplete data and 2 cases of loss to follow-up, 78 patients, including 13 females (16.7%) and 65 males (83.3%) were finally eligible for the study, with the median age of 56 years. There were 52.6% of patients with the tumor size <5 cm, 69.2% with the tumor located in the right lobe, 74.4% with TNM staging I, and 84.6% with HBV/HCV infection. The clinicopathological features and follow-up duration of 78 HCC patients were summarized in Supplementary Table 1.
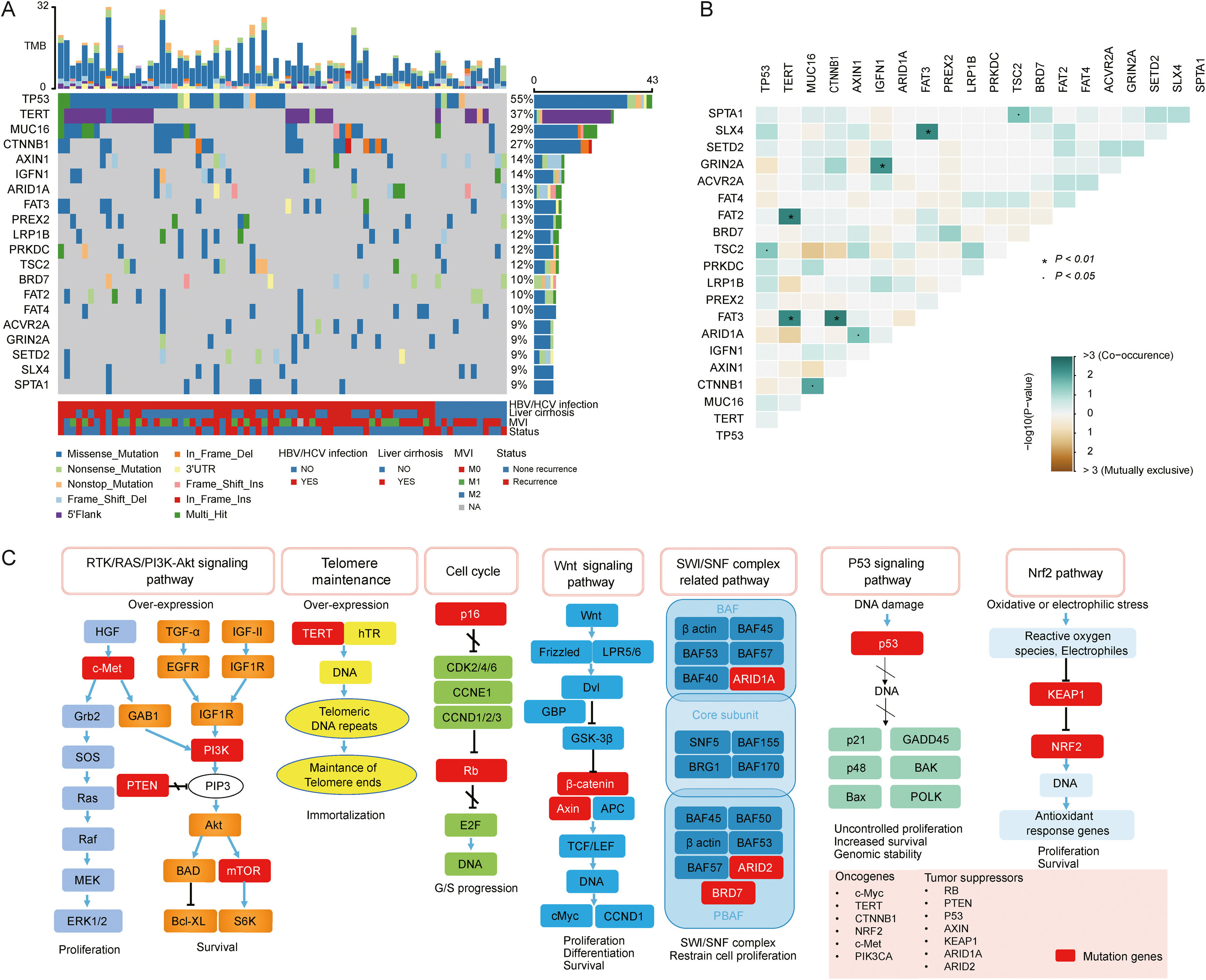
3.2Landscape of genetic aberrations in HCCAmong 78 HCC samples sequenced, it could be observed that TP53 (55%), TERT (37%), MUC16 (29%), CTNNB1 (27%), AXIN1 (14%), IGFN1 (14%), ARID1A (13%), FAT3 (13%) and PREX2 (13%) were commonly mutated in HCC (Fig. 1A). The epistatic interactions of frequently mutated genes occurring ≥10% of all samples showed co-occurrences between TSC2 mutation and TP53 and SPTA1 mutations, TERT mutation and FAT2 and FAT3 mutations, CTNNB1 mutation and MUC16 and FAT3 mutations, ARID1A mutation and AXIN1 mutation, GRIN2A mutation and IGFN1 mutation, as well as SLX4 mutation and FAT3 mutation. (Fig. 1B). The enrichment condition of KEGG pathways in mutated genes was depicted in Fig. 1C.
 A waterfall plot presents the landscape of genomic mutations in HCC, (B) Major epistatic interactions between mutated driver genes occurring ≥10% in HCC, (C) The enrichment condition of Kyoto Encyclopedia of Genes and Genomes pathways in mutated genes based on a hypergeometric test and Bonferroni correction.")
Landscape of genomic aberrations in HCC. (A) A waterfall plot presents the landscape of genomic mutations in HCC, (B) Major epistatic interactions between mutated driver genes occurring ≥10% in HCC, (C) The enrichment condition of Kyoto Encyclopedia of Genes and Genomes pathways in mutated genes based on a hypergeometric test and Bonferroni correction.
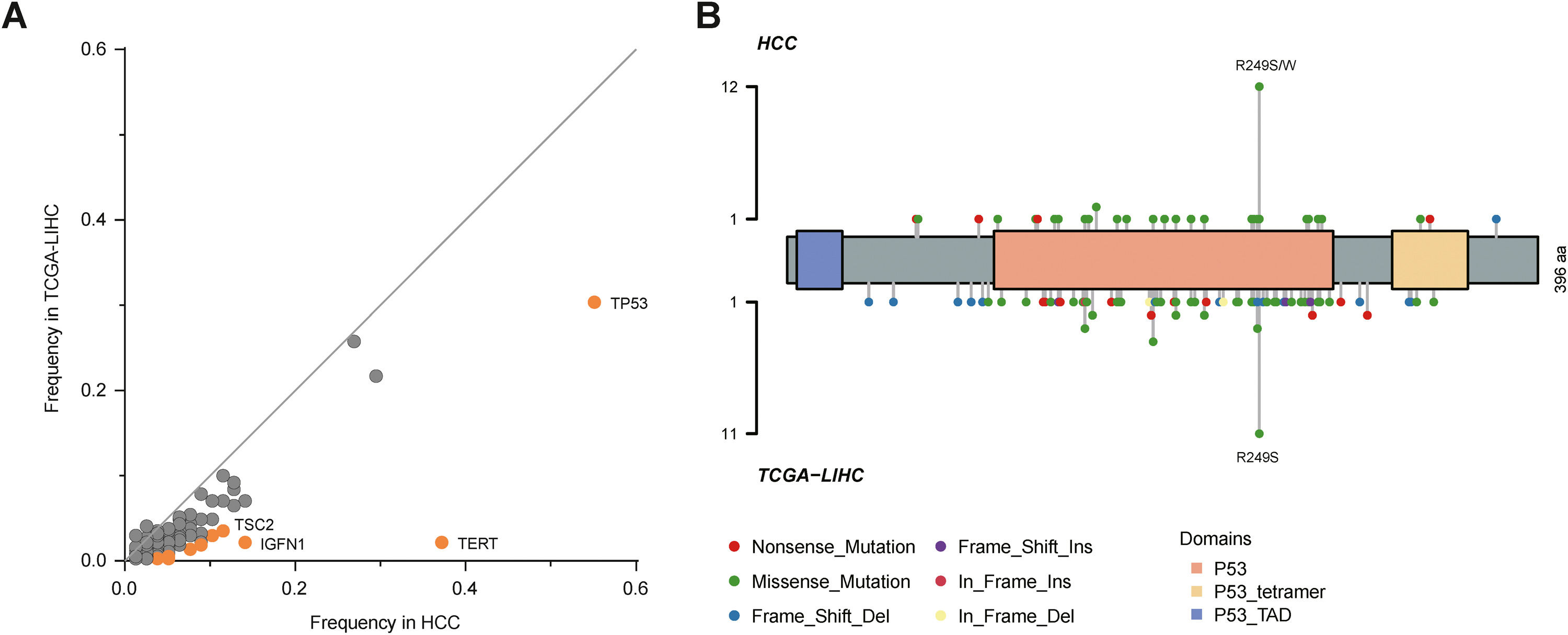
By comparison to the frequencies of mutated genes in our cohort with those in The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) cohort, TP53, TERT, TSC2 and IGFN1 mutations were identified as statistically significant (P<0.05; Fig. 2A), among which TP53 mutation was most significant. Fig. 2B described TP53 mutation sites between our cohort and TCGA cohort. It could be observed that R249S was most common in these two cohorts.
3.3Association of genetic aberrations with clinicopathological features and immunotherapy biomarkers and TP53 mutation sites (B) between our cohort and TCGA cohort. Orange dots represent the genes with significant differences, in which Fisher")
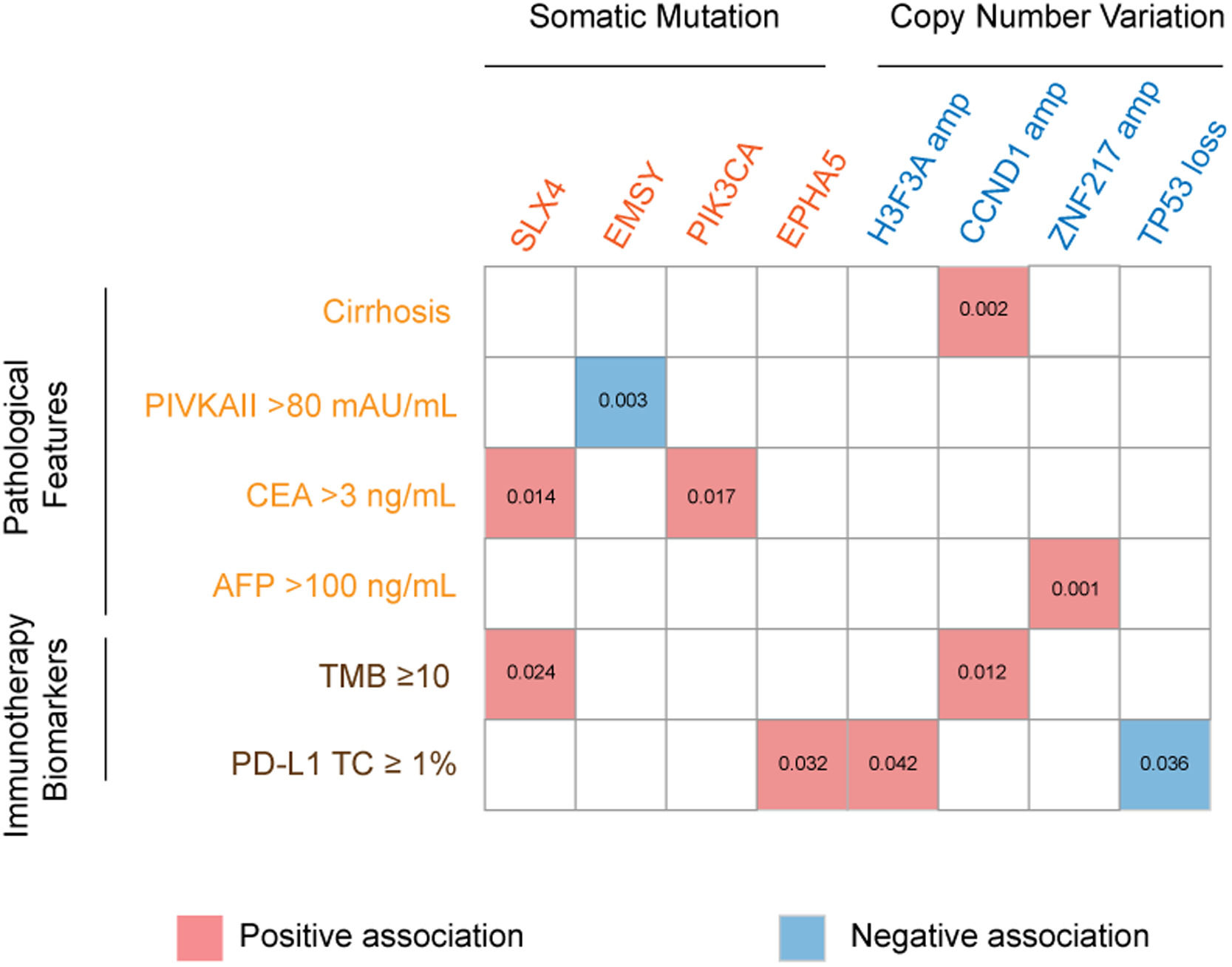
The association of genetic aberrations in HCC with clinicopathological features, tTMB and PD-L1 expression was analyzed (Fig. 3; Supplementary Table 2). As was shown, SLX4 mutations were positively correlated with CEA >3 ng/mL and tTMB-high, while EMSY mutations were negatively associated with PIVKAII >80 mAU/mL. Significantly positive associations were also presented between PIK3CA mutations, CEA >3 ng/mL and EPHA5 mutations, between H3F3A amplification and PD-L1 positive expression, between CCND1 amplification, liver cirrhosis and tTMB-high, as well as between ZNF217 amplification, liver cirrhosis and AFP>100 ng/mL. Additionally, TP53 loss was negatively associated with PD-L1 positive expression.
, carcinoma embryonic antigen (CEA), alpha fetoprotein (AFP), tumor mutation burden (TMB), programmed cell death ligand-1 (PD-L1), tumor cells (TC).")
The association of genomic aberrations in HCC with clinicopathological features, TMB and PD-L1 expression. The value of ≥10 mut/Mb was defined as the TMB-high level, and PD-L1 positivity was defined when the value of PD-L1 positive tumor cells was more than 1. Abbreviations: protein induced by vitamin K absence/antagonist-II (PIVKA-II), carcinoma embryonic antigen (CEA), alpha fetoprotein (AFP), tumor mutation burden (TMB), programmed cell death ligand-1 (PD-L1), tumor cells (TC).
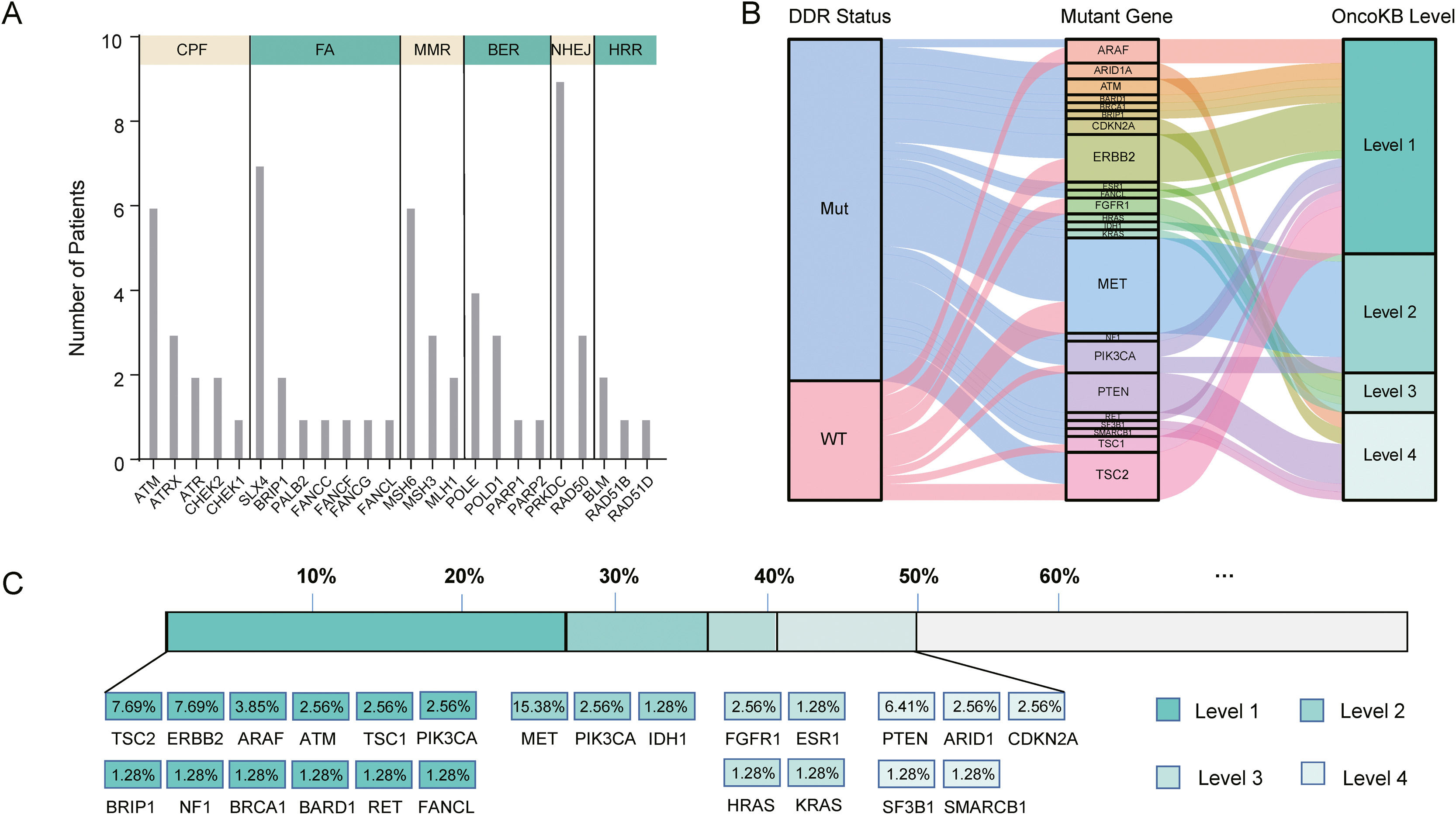
In our cohort, 48 cases (61.5%, 48/78) harbored DDR gene mutations, with most mutations in PRKDC (n=9), SLX4 (n=7), ATM (n=6), MSH6 (n=6), and POLE (n=4) (Fig. 4A). According to the presence or absence of DDR gene mutations, the patients were classified into DDR-mutant and wild-type groups for comparing the drug-matching difference across indications. Among 48 DDR-mutated patients, 31 cases were matched actionable mutations, while only 8 out of 30 DDR wild-type patients were matched actionable mutations. The matched drugs of DDR-mutant patients were significantly more than those of wild-type patients (P=0.0011, Fig. 4B).
 The number of patients with DDR gene mutations in our cohort, (B) The flow diagram represents the list of translational targets in DDR-mutant and wild-type patients for each OncoKB recommendation level, (C) The drug-matching condition across indications in our cohort. Abbreviations: DNA damage repair (DDR), check point factors (CPF), Fanconi anemia (FA), mismatch repair (MMR), base excision repair (BER), non-homologous end-joining (NHEJ), homologous recombination repair (HRR), Mut (mutations), WT (wild type).")
DDR gene mutations and drug-matching conditions. (A) The number of patients with DDR gene mutations in our cohort, (B) The flow diagram represents the list of translational targets in DDR-mutant and wild-type patients for each OncoKB recommendation level, (C) The drug-matching condition across indications in our cohort. Abbreviations: DNA damage repair (DDR), check point factors (CPF), Fanconi anemia (FA), mismatch repair (MMR), base excision repair (BER), non-homologous end-joining (NHEJ), homologous recombination repair (HRR), Mut (mutations), WT (wild type).
As annotated by OncoKB, at least one actionable mutation occurred in 50.0% of patients (39/78), including 26.92% (TSC2, ERBB2, ARAF, ATM, TSC1, PIK3CA, BRIP1, NF1, BRCA1, BRAD1, TET and FANCL) at level 1, 23.08% (MET, PIK3CA and IDH1) at level 2, 17.95% (FGFR1, ESR1, HRAS and KRAS) at level 3 and 14.1% (PTEN, ARID1, CDKN2A, SF3B1 and SMARCB1) at level 4 (Fig. 4C; Supplementary Table 3). Of these 39 patients with actionable mutations, 31 cases harbored DDR gene mutations, while 8 did not.
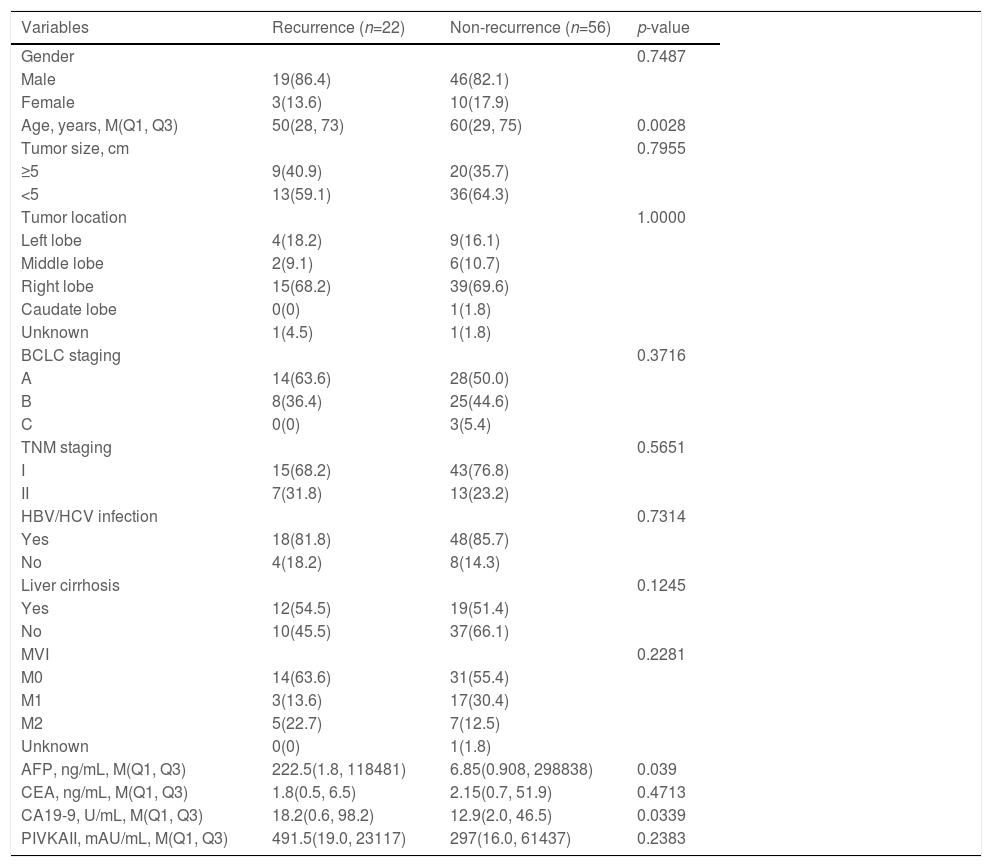
3.5Predictors for postoperative recurrenceOf 78 cases, 22 suffered from recurrence within two years following surgery, while 56 didn't. The difference was pronounced between recurrent and non-recurrent patients in age (p=0.0028), AFP (p=0.039) and CA19-9 (p=0.0339; Table 1). Regarding the association between the top 50 mutant genes and recurrence in HCC, univariate and multivariate analyses both revealed that FH amplification (odds ratio [OR]: 3.752, 95% confidence interval [CI]: 1.170-12.028, p=0.026) and RB1 mutations (OR: 13.185, 95%CI: 1.214-143.198, p=0.034) were the independent risk factors for postoperative early recurrence (Supplementary Table 4; Table 2).
Clinicopathological features of patients with and without postoperative recurrence, n (%).
BCLC, Barcelona clinic liver cancer; MVI, microvascular invasion; HBV, hepatitis B virus; HCV, hepatitis C virus; AFP, alpha fetoprotein; CEA, carcinoma embryonic antigen; CA19-9, carbohydrate antigen 19-9; PIVKAII, protein induced by vitamin K absence/antagonist-II.
Univariate and multivariate models of HCC postoperative early recurrence.
OR, odds ratio; CI, confidence interval.
Precise molecular classification of liver cancer not only contributes to its individualized diagnosis and treatment as well as personalized administration but also to enhance the understanding of clinicians on the complexity and heterogeneity. In this study, we characterized the landscape of genetic alterations in Chinese HCC patients and analyzed the association with clinicopathological features and prognosis. The results showed that TP53, TERT, MUC16, CTNNB1, AXIN1, IGFN1, ARID1A, FAT3 and PREX2 were frequently mutated in HCC, some of which related to the clinicopathological features. In addition, FH amplification and RB1 mutations were associated with an increased risk of early postoperative recurrence in HCC.
The epistatic interaction between mutated driver genes plays a crucial role in determining the carcinogenic process. In HCC, three major clusters of positive epistatic interactions (AXIN1 mutations and ARID1A and RPS6KA3 mutations; CTNNB1 mutations and ARID2, TERT promoter and NFE2L2 mutations; TP53 mutations and KEAP1, TSC2 mutations as well as CCND1/FGF19 amplification) and 1 negative interaction (CTNNB1 mutations and TP53 and AXIN1 mutations) have been reported [4,7,17]. In our study, novel epistatic interactions were identified, such as positive interactions between TERT mutations and FAT2 and FAT3 mutations, CTNNB1 mutations and MUC16 and FAT3 mutations, GRIN2A mutations and IGFN1 mutations, as well as SLX4 mutations and FAT3 mutations. By comparing the frequencies of mutated genes in our cohort with those in TCGA-LIHC, TP53, TERT, TSC2 and IGFN1, mutations were identified to have significant differences, which may be attributed to different populations and tumor stages. In addition, our results confirmed the close relation between genetic aberrations and clinicopathological features, such as positive associations of PIK3CA mutations with CEA >3 ng/mL, and CCND1 amplification with liver cirrhosis and tTMB-high.
In our study, KEGG enrichment analysis revealed that various pathways, such as RTK/RAS/PI3K-Akt signaling pathway, cell cycle, SWI/SNF complex related pathway and Wnt signaling pathway, were significantly enriched for the mutated genes in HCC, which may provide a potential source for new molecular targets of novel therapies. PI3K-Akt signaling pathway is highly mutated and activated in several cancer types [18], and is associated with HCC progression, vascular infiltration and metastasis, as well as poor prognosis [19]. Deregulation of the cell cycle is a leading cause of the unrestricted proliferation of cancer cells. A previous study demonstrated that cell cycle was the most significantly enriched pathway for differentially expressed genes in HCC [20]. ARID1A and ARID2 mutations in SWI/SNF complex related pathway are common in HCC. They both participate in transcriptional activation and suppression of select genes through chromatin remodeling [21]. Notably, in agreement with previous findings [22,23], our results also demonstrated the Wnt signaling pathway was the significantly altered pathway in HCC, highlighting the link between this signaling and hepatocarcinogenesis.
Genomic instability, such as chromosome segregation defects, telomere erosion and aberrations in the DDR pathways, has been a common characteristic of HCC [24–26]. Although various types of hepatocarcinogenesis models have been depicted [27,28], the direct association of epigenetic and genetic alterations with HCC remains unascertained. Multiple HCC-related risk factors have been demonstrated to facilitate DNA damage, formation of DNA adducts and chromosomal alterations, thus aberrations in the DDR pathways may accumulate these lesions to induce hepatocarcinogenesis and to promote advanced HCC progression [29]. DDR gene alterations in cancer can result in homologous recombination deficiency, genomic instability, and high TMB [30]. There is a study suggesting that patients with DDR-mutated primary liver cancer may be reasonable candidates for precision cancer treatment [31]. In our study, 61.5% of HCC patients harbored at least one DDR mutation, significantly higher than 20.3% reported by Lin et al. [31]. This great difference may be explained by the number of DDR genes contained in the NGS-based gene panel. Totally 66 DDR genes were included in our panel, significantly different from 31 DDR genes in the previous study. Moreover, although the direct clinical influence of prospective NGS in HCC had less utility compared with other solid tumors like melanoma and lung cancer [32], 50.0% of DDR-mutated patients in our study showed at least one actionable aberration that might be a potential target for currently available drugs approved by Food and Drug Administration or agents in active clinical development. These findings may be of great importance for the translational significance of clinical treatment using immune checkpoint inhibitors or PARP inhibitors.
The FH gene is located at the chromosome locus 1p43 in the human genome [33]. Loss of FH function is correlated with metabolic dysregulation, which can result in cell survival via a pseudohypoxia response and aerobic glycolysis. FH mutations can alter the migratory capability of tumor cells, responses to oxidative stress and DNA damage [34]. Recently, FH mutations have been reported to involve in the pathogenesis of various cancer types, including hereditary leiomyomatosis and renal cell cancer, glioma, breast cancer, etc. [34–36], suggesting a key role of FH loss in human cancers. Zhang et al. found that FH-deficient renal cell carcinoma was associated with poor prognosis, and detection of FH mutations was conductive to the tumor diagnosis [37]. For FH-deficient renal cell carcinoma patients with suspicious clinical or pathological characteristics, the detection of FH mutations should be taken into consideration [38]. In our study, analysis of the association between genomic characteristics of HCC and recurrence first revealed that FH amplification was an independent risk factor for early postoperative recurrence in HCC. However, this association needs to be further clarified in large-scale, well-designed studies.
The RB1 gene, a tumor suppressor gene, is frequently silenced in various human cancer types, including HCC [39]. Although the pRb protein is usually downregulated in HCC cells, RB1 mutations are relatively uncommon in HCC [39,40]. By contrast to the early-stage HCC (BCLC staging A), advanced-stage HCC (BCLC staging B/C) indicated a higher frequency of RB1 mutations (14.6% vs. 2.6%) [17]. In the present study, the frequency of RB1 mutations in TNM staging I-II HCC patients was 6.4%. The difference in RB1 mutations between these two studies may be attributed to the use of different HCC staging and study populations. Moreover, RB1 mutations were identified as a significant risk factor for the early recurrence of HCC. Unlike our results, Nault et al. found that CDKN2A mutations were independently associated with an increased risk of tumor recurrence [17]. This may be explained by disparate study populations and sequencing assays.
Although this was a retrospective study with a small sample size and some potential influencing factors for postoperative recurrences, such as AFP levels, may be underestimated, but there were still several strengths that should be underlined. The major strength of our study was that it first demonstrated the association of FH amplification and RB1 mutations with early postoperative recurrence in HCC. Second, most patients included in our study were males, accounting for 83.3% of the study population. This was similar to the male proportion of previous studies [17,41]. Additionally, novel interplays between mutated driver genes, like positive interactions between TERT mutations and FAT2 and FAT3 mutations, CTNNB1 mutations and MUC16 and FAT3 mutations were identified. Due to the absence of explicit oncogenic addiction loops in HCC, a comprehensive understanding of the interplay between mutated driver genes may contribute to the development of more potential combination-based therapies.
5ConclusionsOur study provides a novel insight into the genomic profiling of Chinese HCC patients and shows the correlation between the genetic variants and clinicopathological features. FH amplification and RB1 mutations may be associated with a higher risk of early postoperative recurrence in HCC. In view of the small sample size in our study, the association of FH amplification and RB1 mutations with HCC recurrence still needs to be investigated in more large-scale, well-designed studies.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author contributionsZ. Yang, J.W. Liu, and F. Xue: conceptualization and writing-original draft preparation; L. Zhang, H. Xue, Y.Y. Wu, and S.L. Bai: data collection and methodology; X.J. Hu, F.R. Du, X.X. Wang, and W.L. Deng: data curation, formal analysis and investigation; C. Song and K. Wang: conceptualization, writing-reviewing and editing, supervision.
Data availability statementThe data that support the findings of this study are available from the first author and corresponding author upon reasonable request.



