




Autoimmune pancreatitis (AIP) is a unique form of inflammatory disorders that affect this organ.1,2 Histopathology reveals dense infiltration of T lymphocytes and IgG4-positive plasma cells with fibrosis and obliterative phlebitis, termed lymphoplasmacytic sclerosing pancreatitis (LPSP).1–3 Various extrapan-creatic lesions associated with AIP have similar his-tological features.4,5 Therefore, AIP is currently considered a pancreatic manifestation of an IgG4-re-lated systemic disease. In some cases, there is clinical involvement of only 1 or 2 organs, while in others 3 or 4 are affected.2,5,6 However, precise pathogenetic mechanisms, including the role of IgG4, remain unclear.
Stenosis of the bile duct is frequently observed in AIP patients; histologically, IgG4-related sclerosing cholangitis, a bile duct lesion of the systemic disease, is revealed.2,6 Stenosis is seen most frequently (70-79%)7,8 in the lower bile duct of AIP patients, but this may be due in part to compression caused by pancreatic edema, in addition to biliary wall thick-ening.9 When stenosis develops in the hilar or intra-hepatic bile duct, the cholangiographic appearance is similar to that of primary sclerosing cholangitis (PSC).7,8 PSC is a progressive disease that eventually involves intra- and extra-hepatic bile ducts, and which sometimes leads to liver cirrhosis.10,11 The value of steroid therapy is questionable, and liver transplantation is the only effective curative treatment. Since IgG4-related sclerosing cholangitis responds well to steroid therapy, it is necessary to differentiate this from PSC in order to provide the most appropriate treatment regimen.
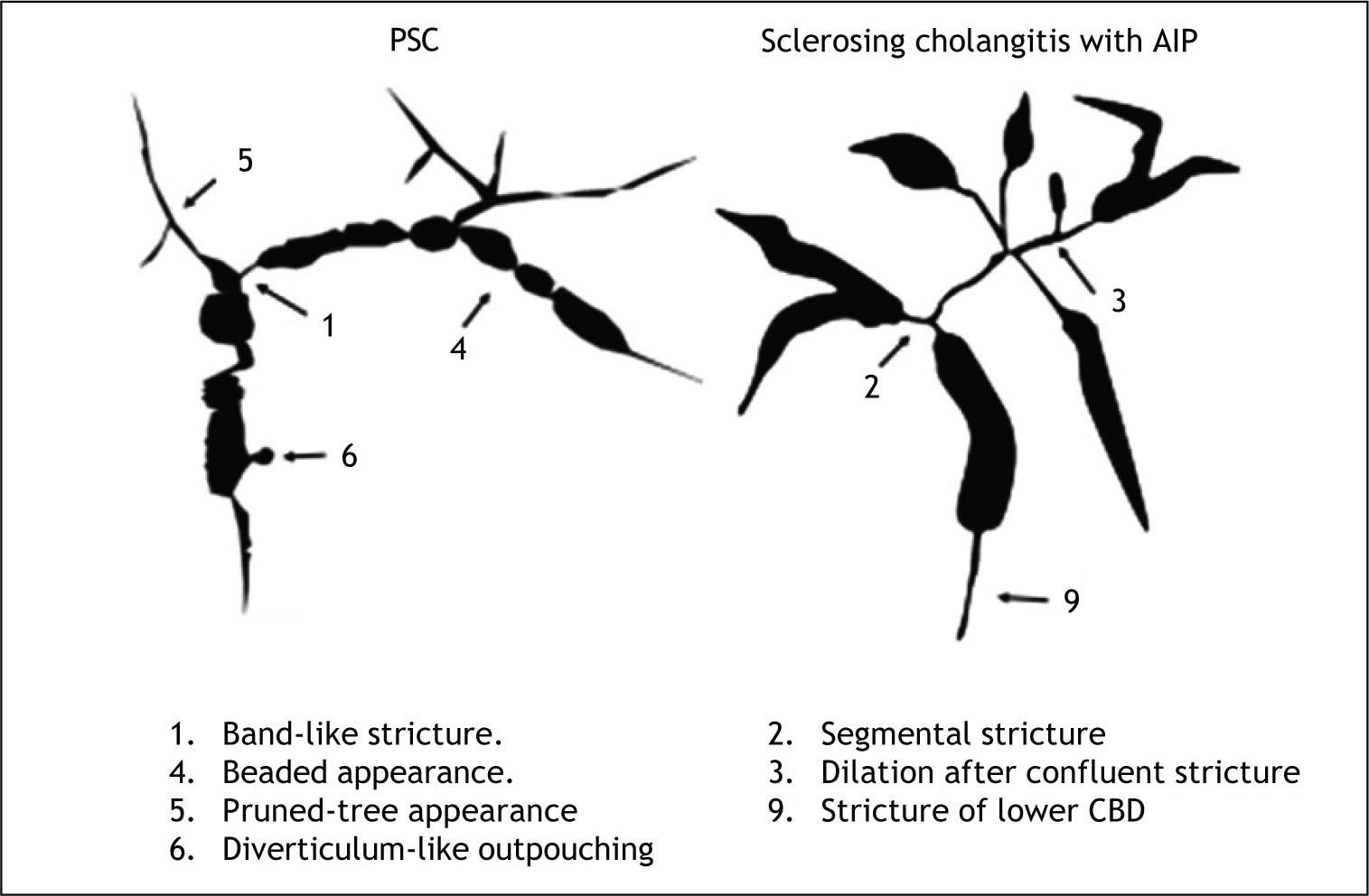
IgG4-related sclerosing cholangitis and PSC can be discriminated clinically, serologically, radiologically, and histopathologically. Average age of clinical onset for IgG4-related sclerosing cholangitis is approximately two decades older than for PSC, which occurs during in 30 to-40-year-olds.7,8,12 Obstructive jaundice is the most frequent chief complaint, seen in 75-77%13,14 of patients with IgG4-related sclero-sing cholangitis, whereas many PSC patients are asymptomatic, presenting after liver injury is identified on routine physical examination. Inflammatory bowel disease is sometimes comorbid with (41-63%),13,14 while various sclerosing lesions (such as sclerosing cholecystitis, sclerosing sialadenitis, and retroperitoneal fibrosis) are sometimes seen in pa-tients with IgG4-related sclerosing cholangitis. Serum IgG4 levels are frequently and significantly elevated in patients with IgG4-related sclerosing cholangitis7,8,13,14(Table 1). Mendes, et al.15 reported elevated serum IgG4 levels in 9% (12 of 127) of PSC patients, who experienced a more severe disease course, including shorter time to liver transplantation. However, some may have had IgG4-related sclerosing cholangitis without pancreatic involvement. According to Nakazawa’s classification16 of cholangiograms in IgG4-related sclerosing cho-langitis, type 1 stenosis, located exclusively in the lower part of the common bile duct, can often lead to a misdiagnosis of pancreatic cancer; type 2 stenosis, diffusely distributed throughout the intra- and ex-trahepatic bile ducts, is similar to that seen in PSC; while it is type 3 stenosis, of the hilar hepatic region and the lower part of the common bile duct, and type 4 stenosis, detected only in the hilar hepatic region, that should be differentiated from cholangio-carcinoma. As revealed by cholangiogram, IgG4-rela-ted sclerosing cholangitis involves lower bile duct stenosis and a relatively long stricture extending from the hilar to intrahepatic biliary system with simple distal dilatation, whereas PSC produces characteristic findings, including band-like stricture, beaded appearance, pruned tree appearance, and diverticulum-like outpouching16(Figure 1). In IgG4-related sclerosing cholangitis, intraductal or endos-copic ultrasonography can detect wall thickening of intra- or extrahepatic bile ducts in areas without stenosis on cholangiogram.8,17 On pancreatography, patients with IgG4-related sclerosing cholangitis frequent show irregular narrowing of the main pancreatic duct which is characteristic finding of AIP, but pancreatograms of PSC patients are usually normal. As the histological findings of IgG4-related scle-rosing cholangitis include transmural fibrosis and dense lymphoplasmacytic infiltration of the bile duct wall as well as periportal areas of the liver, positive abundant IgG4 immunostaining of bile duct or liver biopsy specimens supports this diagnosis.18 However, the positive rate was not high due to the small specimens.17 Positive IgG4 immunostaining of biopsy specimens taken from the major duodenal papilla is useful to support the diagnosis of AIP or IgG4-related sclerosing cholangitis.19,20 Since, unlike PSC, IgG4-related sclerosing cholangitis responds dramatically to steroid therapy, rapid steroid responsiveness reassures by confirming the diagnosis. If a diagnosis cannot be obtained using the procedures described above, a steroid trial may be useful. However, to avoid delaying necessary surgery, a short-term (usually 2 weeks)21 trial should be performed carefully, and only by clinicians specializing in pancreatic and biliary disease. Routine steroid trials conducted by general practitioners should be strongly discouraged.
Differences between IgG4-related sclerosing cholangitis and primary sclerosing cholangitis (PSC).
| IgG4-related sclerosing cholangitis | PSC | |
|---|---|---|
| • Age | Elderly | Young |
| • Initial symptom | Obstructive jaundice | Liver dysfunction |
| • Associated diseases | ||
| ° Inflammatory bowel disease | Rare | Sometimes |
| ° Sclerosing diseases | Frequent | Rare |
| • Elevation of serum IgG4 | Frequent | Rare |
| • Infiltration of IgG4-positive cells | Many | Scarce |
| • Steroid responsiveness | Good | Poor |
| • Prognosis | Good | Progressive |

In this issue, Clendenon, et al.22 report a 54-year-old female who twice underwent orthotopic liver transplant for recurrent sclerosing cholangitis after pancreaticoduodenectomy performed for AIP on suspicion of pancreatic cancer. Because the correct diagnosis was not made for 11 years, she did not have steroid therapy until receiving the second liver transplant. Surprisingly, her explanted liver allograft revealed many IgG4 positive plasma cells similar to those seen in her native liver. We suggest that this case demonstrates the natural course of IgG4-related sclerosing cholangitis. Although the prognosis of this disease is generally good due to good steroid responsiveness, its natural behavior may be aggressive, leading to progressive end-stage liver cirrhosis, if appropriate therapy is not administered. In contrast, Koyabu, et al.,23 recently reported a series of PSC cases having elevated serum IgG4 levels and infiltration of abundant IgG4-positive plasma cells in the hepatic portal area who did not respond to steroid therapy.
Recently, IgG4-associated autoimmune hepatitis (AIH) has been reported. Umemura, et al.24 proposed provisional diagnostic criteria for this entity, as follows:
- •
Definitive presence of AIH, according to the International Autoimmune Hepatitis Group (IAI-HG) scoring system.
- •
Serum IgG4 concentration > 135 mg/dL.
- •
Abundant infiltration of IgG4-positive plasma cells in the portal tract.
Using these criteria, they found 2 IgG4-associated AIH patients among 60 AIH patients. Interestingly, one such patient who was given low-dose steroid therapy developed IgG4-related sclerosing cholangi-tis after 5 years of follow-up. An AIH/PSC overlap syndrome has been reported,25 and this patient might fall into that category.
Further study is needed to clarify the true nature of IgG4-related sclerosing cholangitis.



