



The use of immunosuppressive medications for solid organ transplantation is associated with cardiovascular, metabolic, and oncologic complications. On the other hand, the development of graft rejection is associated with increased mortality and graft dysfunction. Liver transplant recipients can withdraw from immunosuppression without developing graft injury while preserving an adequate antimicrobial response - a characteristic known as immunotolerance. Immunotolerance can be spontaneously or pharmacologically achieved. Contrary to the classic dogma, clinical studies have elucidated low rates of true spontaneous immunotolerance (no serologic or histological markers of immune injury) among liver transplant recipients. However, clinical, serologic, and tissue biomarkers can aid in selecting patients in whom immunosuppression can be safely withdrawn. For those who failed an immunosuppression withdrawal trial or are at high risk of rejection, pharmacological interventions for immunotolerance induction are under development.
In this review, we provide an overview of the mechanisms of immunotolerance, the clinical studies investigating predictors and biomarkers of spontaneous immunotolerance, as well as the potential pharmacological interventions for inducing it.
Liver transplantation (LT) is the most effective treatment for end-stage liver disease, fulminant acute liver failure, and selected cases of liver cancer [1]. Advances in immunosuppressive therapies have led to a drastic increase in LT survival rates [2]. However, chronic immunosuppression (IS) often leads to metabolic, cardiovascular, renal, and oncologic complications that negatively impact long-term survival and quality of life [2,3].
In contrast to other organs, LT recipients can maintain low levels (or even withdraw) of IS without developing graft injury—a characteristic known as immunotolerance. Immunotolerance can be either deletional or regulatory. Deletional tolerance refers to negative selection for auto-reactive T-cells. In the context of liver transplantation, the deletion of auto-reactive and graft-reactive T-cells is called "mixed-chimerism" [4]. This process leads to graft tolerance while preserving the immune armamentary to fight infections [5–7]. Regulatory tolerance is mediated by T-cells that successfully underwent negative selection and developed immunomodulatory properties; the T regulatory cells (Treg). Treg cells downregulate reactions against self, and in the case of LT, against graft [8–10].
Up to 20-40% of LT recipients can maintain graft tolerance after ceasing IS [11]; this is known as spontaneous operational immunotolerance (SOI). In addition, Immunotolerance can be pharmacologically induced, a concept known as therapeutic operational immunotolerance [12]. Identifying risk profiles for successful IS withdrawal and developing pharmacological interventions for those at high risk for rejection is the holy grail of immunotolerance research.
The following narrative review, targeting the practicing gastroenterologist/hepatologist, aims to discuss the current concepts of immunotolerance in LT.
2What's to gain? The delicate balance between toxicity and rejectionThe development (or worsening) of metabolic syndrome is a well-known complication of IS [13]. After LT, there is a significant increase in the prevalence of hypertension (from 15% to 70%) [14], type 2 diabetes mellitus (15% to 40%) [15,16], dyslipidemia (up to 70% of LT recipients) [16], obesity [17], and chronic kidney disease (>50% will have stage 3 in 2 years after LT and 20% stage 4 in 5 years) [18,19]. Consequently, the cardiovascular risk increases substantially, becoming a leading cause of mortality and morbidity among recipients (up to 20%) [20–22]. Although developing metabolic syndrome is a multifactorial event, the temporal association between IS withdrawal and improved cardiovascular risk, metabolic profile [23], and kidney function [24] suggests a causal relationship.
In LT recipients, lymphoproliferative and solid organ malignancies occur two and 30-fold more frequently compared to the general population [25]. Dose and duration of IS are major risk factors [26,27]. In these cases, IS reduction [28] or withdrawal [29] is usually attempted as a salvage intervention to reestablish an anti-tumoral immune response. Although speculative, withdrawing IS could potentially serve as primary prevention for de novo malignancies and recurrences.
On the other hand, the metabolic and oncologic benefits must be weighted with the risk of rejection and allograft dysfunction. As older studies suggested no association with graft failure or mortality, LT was traditionally considered exempt from the deleterious effects of acute rejection [30,31]. However, data from the A2ALL cohort demonstrated a significant increase in graft failure and all-cause mortality in those who developed acute rejection [32]. To be noted, this study did not consider chronic subclinical allograft injury (biopsy-proven injury without serologic markers of damage). Therefore, the clinical implications of the latter remain unknown.
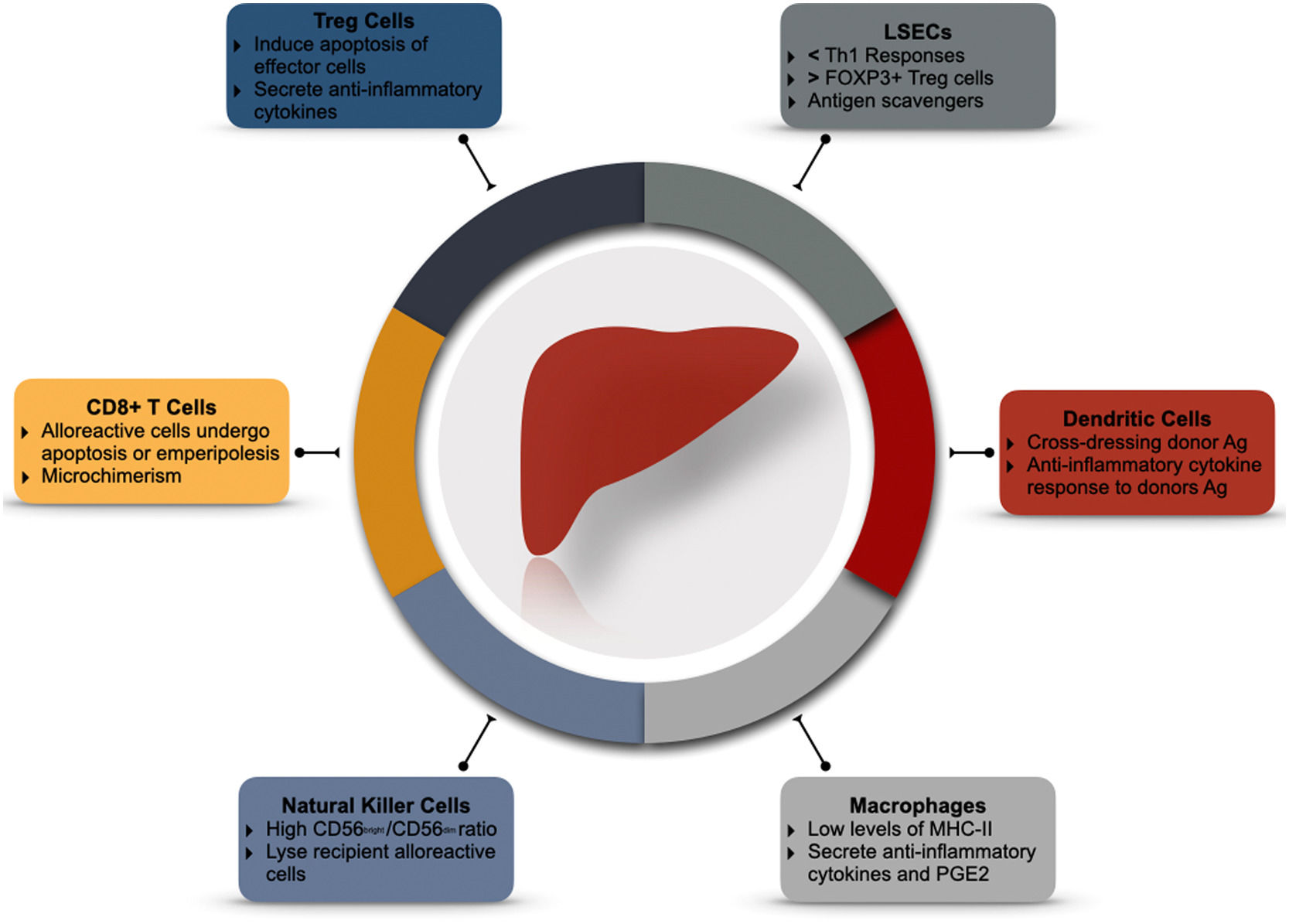
3Back to the basics: why is the liver immunotolerant?3.1The liver has it: liver architecture and microenvironmentBy receiving 75% of its blood flow through the portal circulation, the liver is constantly exposed to a myriad of environmental antigens from the gastrointestinal tract and spleen [33]. Therefore, it must remain vigilant for pathogenic bacteria and malignant cells while tolerating harmless antigens. When blood passes through the liver sinusoids, it enters the subendothelial Disse space through the fenestrated liver sinusoidal endothelial cells (LSECs). This low flow environment favors antigen exposure of blood contents with the hepatic reticuloendothelial system; LSECs, Kupffer cells (KC), dendritic cells (DC), intrahepatic lymphocytes, and other hematopoietic cells [34,35] (seeFig. 1).

LSECs are a peculiar group of cells that act as atypical antigen-presenting cells (APC). Although they present antigens to CD4+ and CD8+ T-cells, they lack the capacity for IL-12 expression and, therefore, cannot mount a robust TH1 response [36]. On the contrary, their interaction with CD4+ and CD8+ T-cells induces the differentiation of FOXP3+ Treg cells [37] and upregulation of programmed death ligand-1 (PDL-1) [38] signaling. LSECs can also induce T-cell apoptosis via the Fas-FasL pathway, favoring the immune-tolerant recognition of antigens. Experimental models have evidenced the role of specific LSECs proteins such as LSECtin. After the induction of acute liver injury, LSECtin knockout mice develop higher T-cell activation and accelerated liver damage than controls. A response blunted by recombinant LSECtin [36]. LSECs and KC constitute the most potent scavenger system in the body. They recognize phagocyte microbial-associated molecular patterns (MAMPs) and damage-associated molecular patterns (DAMPs) arising from the gut. Filtering these contents dampens the inflammatory response to exogenous antigens [39]. LSECs favor a selective, attenuated immune response to antigens.
3.2The dendritic cell: a bridge to toleranceDCs are classic APCs involved in antigen uptake, processing, and presentation. They orchestrate the interactions between innate and adaptative immunity, which has placed them in the spotlight as potential therapeutic targets for inducing immunotolerance [40–43].
The liver recruits hematopoietic precursors and monocytes to differentiate into a regulatory DC. These so-called conventional DC (cDC) express low levels of major histocompatibility complex class II (MHC-II) but high levels of PD-L1, CD39, and DAP12 [34–44]. After cDCs are exposed to gut-derived antigens, the secretion of pro-inflammatory cytokines (IL-12) diminishes, while anti-inflammatory cytokines (IL-10, IL-27, and TGF-β) are favored. The result is the development of Tregs and blunting of effector T-cells activation [34]. Interestingly, the in-vivo infusion of dexamethasone mimics their effects [44]. In addition, DCs can acquire donor MHC antigens shortly after transplantation and expose them to T cells [45,46]. This "cross-dressing" of DCs with donor antigens regulates anti-donor T-cells and promotes graft tolerance [47].
The second group, non-conventional plasmacytoid dendritic cells (pDCs), are APCs and Treg enhancers. However, they have antiviral activity (through IFN-gamma and T-cell modulation) and similar genotypes as T and B lymphocytes [48]. In experimental models of MHC-mismatched LT, pDCs depleted mice have reduced graft survival compared with pDCs treated mice [49].
The disruption DCs mediated interactions have shed light on possible therapeutic interventions. The interruption of PD-1/PD-L1 signaling through KO models or anti-PD-L1 antibodies resulted in allograft rejection and increased inflammatory markers [48]. Similarly, a model of ischemia-reperfusion injury demonstrated worsened liver injury in cDC depleted mice. This effect was attenuated by cDC's anti-inflammatory cytokines (IL-10) [41] and cDC/Treg cell infusion. Currently, multiple trials are evaluating the safety of donor-derived cDC for immune tolerance induction (See Inducing Tolerance)
3.3Macrophages: more than just engulfersMacrophages play an essential role in phagocytizing exogenous and endogenous antigens. There are two main subpopulations of liver macrophages; KC, which are liver resident macrophages that express low concentrations of MHC-II and co-stimulatory molecules. They secrete anti-inflammatory cytokines (IL-10 and TGF-β) [50,51] and inhibit T-cell activation [52]. On the other hand, monocyte-derived macrophages arise from circulating cells and can infiltrate and initiate an inflammatory response after liver injury [53].
In LT, donor-derived KCs are responsible for the initial response to ischemia/reperfusion injury and repair. Zhao et al. demonstrated the role of KC in inhibiting acute allograft rejection by modulating inflammatory responses through IL-34 [54]. On the other hand, the recipient's monocyte-derived macrophages infiltrate the liver after the initial injury and mediate allograft rejection responses [55].
3.4The "Other" innate immune cellsNatural Killer cells (NK) represent half of the intrahepatic lymphocyte population. They recognize autologous cells through MHC-I receptors exposing self-antigens [55]. In the absence of these antigens, NK cells initiate a robust cytolytic response [56,57]. Liver NK cells have a more reactive phenotype than their peripheral counterpart. They express a higher concentration of activation receptors (CD19, NKp44, NKp46), apoptosis (TRAIL) ligands, and abundant cytotoxic granules [58,59]. Even in the absence of graft-reactive T and B cells, NK cells are primary mediators of graft rejection [57,59].
However, the association between recipient NK gene expression/cell expansion and graft survival is not straightforward. CD56bright NK cells are precursors cells that express CD56 but lack CD16; they have a low cytotoxic potential and cannot mount antibody-dependent cellular toxicity. This subpopulation has an essential role in the induction of tolerance. CD56dim NK cells express CD56 and CD16 and have potent direct and antibody-induced cytotoxic activity [60]. Donor NK cells are thought to be promoters of immune tolerance (either by mounting a direct anti-inflammatory response or by lysing recipient inflammatory cells) and recipients' NK cells as mediators of rejection [55]. Pilot trials are currently testing the feasibility and safety of donor NK cell infusion to prevent hepatocellular carcinoma recurrence after LT [61]. However, their potential role as tolerance inductors is still unexplored.
3.5Acquired immunityThe liver acts as a secondary lymphoid organ by exposing T cells to antigens and deleting those that become auto-reactive [62]. Experimental models have demonstrated that circulating naive CD8+ T cells enter the liver and expose liver antigens. They further develop liver-specific retention markers that are thought to play a role in liver immunosurveillance [63,64]. However, when activated in the allograft, alloreactive CD8+ cells can be eliminated by apoptosis, functional exhaustion, or even undergo suicidal emperipolesis [65–67]. The remaining reactive CD8+ cells develop a poor cytolytic function and effector activity [68]. Worth noting is that the load of the antigen exposure primarily determines CD8+ cells' fate. High loads lead to cell exhaustion and lower loads to effector responses [69]. During LT, there is an exchange between donor lymphocytes (commonly known as passenger lymphocytes) and the recipient, a process known as microchimerism. Microchimerism has been proposed as a tolerance-inducing feature that allows hyporesponsiveness toward the graft. [34,70–72].
On the other hand, Tregs are immune-modulatory cells that can arise from the thymus or the periphery. These cells can suppress innate and adaptative immune responses by expressing inhibitory molecules (ex. CTLA4), inducing apoptosis of effector cells, or releasing inhibitory cytokines (L-10 and TGF-β) [73–75]. Specific phenotypes of Tregs are associated with graft tolerance. Li et al. demonstrated that CD4+CD25+FOXP3+ Tregs are essential for the induction of tolerance (Specifically, the ratio of Tregs to effector cells) [75]. However, their role in maintaining it is uncertain, as CD4+CD25+FOXP3+ Tregs counts decrease steadily through time, and their pharmacological blockade does not seem to impact long-term graft survival [76].
4Who will become immune-tolerant?The decision for IS withdrawal is usually a balancing act between the potential (or ongoing) risks of immunosuppression versus the risk of graft rejection—a decision with critical implications that requires a tailored approach (seeFig. 2). The occurrence of graft tolerance was initially recognized in observational studies in which patients were withdrawn from IS due to IS toxicity, noncompliance, or malignancy. The definition of graft stability was based on serologic parameters of liver injury (elevated transaminases) [77–79]. However, surveillance biopsies from the Kyoto cohort revealed higher fibrosis in patients in whom immunosuppression was withdrawn (despite normal transaminases) [80]. This emphasized the importance of detecting subclinical evidence of graft damage after IS withdrawal. Currently, pre and post-histological evaluation of the graft is recommended. Normal serum markers of liver injury are not considered enough for assuming tolerance [81].

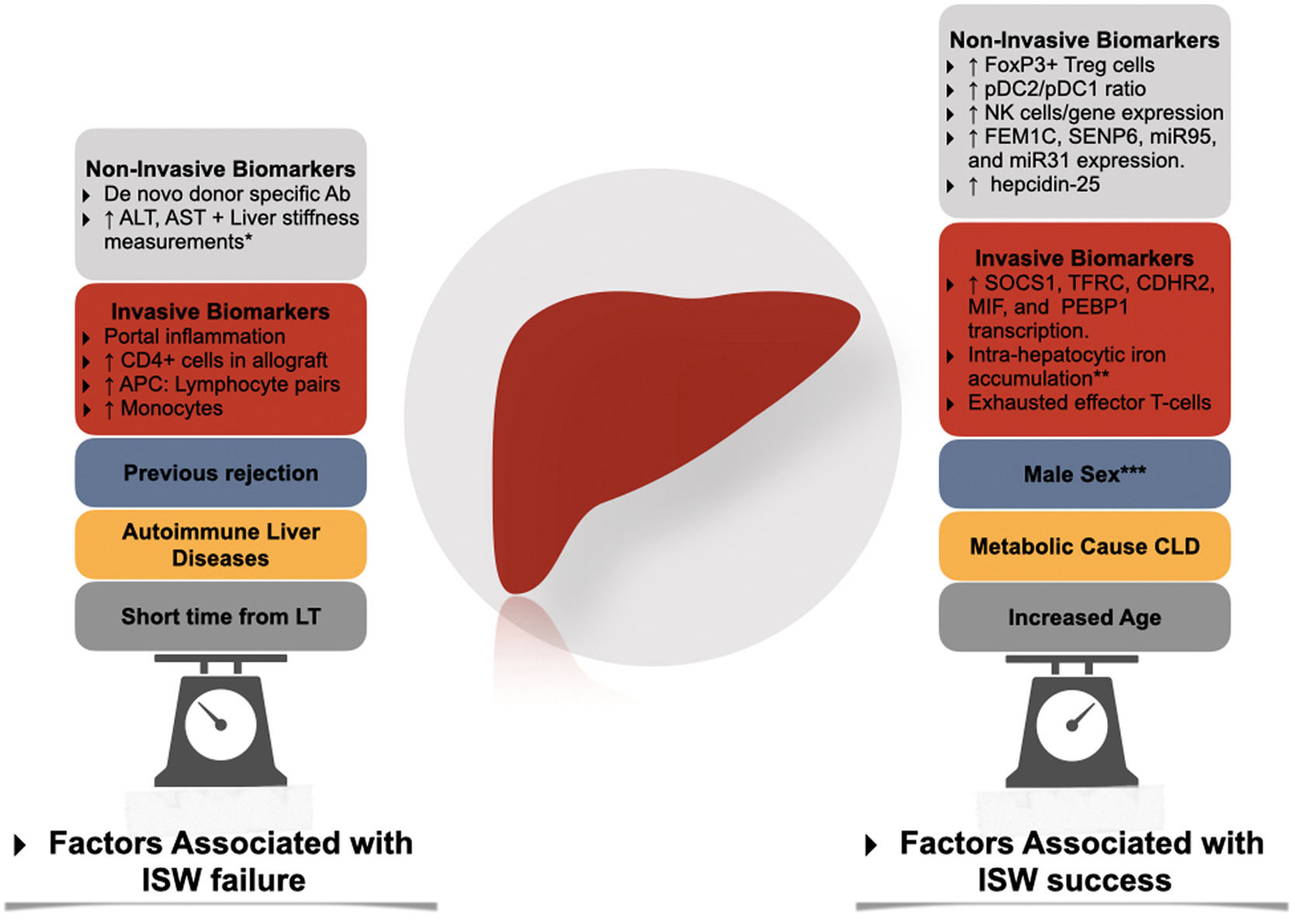
Factors associated with immunosuppression withdrawal failure and success. Ab, antibody; APC, antigen-presenting cells; CLD, Chronic liver disease; ISW, immunosuppression withdrawal; LT, liver transplantation; NK, natural killer; pDC, plasmacytoid dendritic cells. * Through transient elastography-based liver stiffness measurements. ** In conjunction with high serum levels of ferritin and hepcidin-25. *** In adult liver transplantation.
Immune tolerance rates have been reported as high as the 70–80% range in selected populations. However, definite tools to identify the ideal candidate for IS withdrawal are lacking [82].
Two prospective cohorts studied the association of IS withdrawal with the development of tolerance. Both trials excluded populations at high risk for rejection, such as autoimmune liver diseases [32,83], subjects within three years from LT, acute or chronic rejection, and METAVIR III-IV scores in screening biopsy. After three years of follow-up, 40% of the European cohort tolerated IS cessation [83] (tolerance is defined as the absence of biochemical and histological evidence of rejection after 12 months of IS cessation). The other 55% of non-tolerant subjects presented mild rejection episodes that resolved within six months with increased IS. Four patients were withdrawn from the study due to intolerance during IS reduction. Tolerance was independently associated with advanced age at LT and a longer time since LT. Patients with > 10.6 years after LT had a 78% probability of successful withdrawal of IS; this dropped to 38% in patients with more than 5.7 years (but less than 10.6) and 0% in patients with less than 5.7 years and over 49 years old. This data suggests that a state of immunosenescence might favor the development of tolerance as early as in the sixth decade [83,84]. To date, the recipient's age and the interval time from LT to IS withdrawal are the most powerful predictors for the development of tolerance.
A trial performed in 7 transplant centers in the US randomized in a 4:1 ratio withdrawing or maintaining IS after 1-2 years of LT. They excluded previous organ transplants, hepatitis C virus (HCV)+ donors, hepatitis B virus (HBV), hepatocellular carcinoma stage III or higher in the explant, autoimmune etiologies, and significant renal, cardiovascular, or cerebrovascular diseases. Subjects required a control biopsy within three months of randomization without evidence of moderate or severe rejection. A post-withdrawal biopsy was also performed. After a slow withdrawal (scheduled for a year) and two years of follow-up, 67% (52/77) of the subjects had a ≥ 50% reduction in the IS dose, and only 13% (10/77) discontinued IS. Of these, only 19% (10/52) developed tolerance. A complex composite outcome (death, graft loss, severe opportunistic infection or secondary malignancy, Ishak stage ≥ three fibrosis or a > 25% decrease in glomerular filtration rate at 24 months) was higher in the maintenance group (4/13) than in the withdrawal group (12/66). [82]
These studies taught us several lessons: the high selectivity in these cohorts suggests that only a limited amount of patients could benefit from this approach in a real-life scenario. In the European cohort, only 20% (102/500) of the patients screened were enrolled for IS reduction, similar to the USA trial 19% (52/286). Although rejection cases were mild and successfully managed overall, the long-term effects of those findings or the possibility of self-resolution are unclear and likely to remain unanswered. Fewer than 10% of the screened subjects were found to be biopsy-proven tolerant, a number that should make us reconsider if patients with normal serologic markers are truly immunotolerant. Nonetheless, these numbers contrast with those from pediatric studies where higher rates of biopsy-confirmed immune tolerance were achieved [85].
Currently, the OPTIMAL trial (Evaluation of Donor Specific Immune Senescence and Exhaustion as Biomarkers of Tolerance Post Liver Transplantation) [86] and the Liver Immunosuppression Free Trial (LIFT) [87] aim to integrate the results from previous studies by selecting the time from transplantation depending on age (> 50 years three years after LT and < 50 years >6 years) and harmonizing their inclusion/exclusion criteria. These trials aim to collect an extensive biorepository for biomarker development focused on detecting the signature of tolerant hosts.
In the particular case of HCV + LT recipients, the fine tunning between maintaining an adequate anti-viral response and avoiding an immune attack on the graft has made IS withdrawal a controversial matter [88]. Therefore, this group of patients has been typically excluded from large trials. Merrit et al. provided mechanistic explanations supporting the association between immune exhaustion of CD8+ HCV-specific cells and the development of graft tolerance [89]. In this subgroup, HCV status and timing of eradication may prove relevant when deciding on IS withdrawal.
5Biomarkers: can we predict tolerance?Based on the aforementioned trials, the clinical parameters for detecting the "ideal" candidate for safe IS withdrawal are weak (except for time from LT and IS withdrawal) [83]. This led to the search for biomarkers that might assist the clinician in selecting patients for IS withdrawal, stratifying their risk (therefore the optimal timing and strategy), and monitoring for failure [90].
From peripheral cell phenotyping with flow cytometry to gene expression, genomic microarrays, and proteomics in allograft tissue, many serologic markers have been associated with immune tolerance in observational studies and experimental models [34]. Only a few have been tested to predict tolerance prospectively.
6Non invasive biomarkersThe presence of peripheral cells signatures such as a high proportion of FoxP3+ Treg cells [91,92], Treg/Th17 subpopulations [93], Vδ2-TCR γδT cells [94] and a tolerogenic pDC2/pDC1 subset ratio9 have been identified as potential biomarkers predicting operational tolerance. Similarly, in the Barcelona trial, the expression of NK-related genes predicted operational tolerance before IS withdrawn [95], just as a higher proportion of NK-Cells did so in the Pamplona trial [84,96]. The expression of genes (in peripheral cells flow cytometry) such as FEM1C, SENP6, miR95, and miR31 [97,98] has also been demonstrated to predict tolerance. Nonetheless, the predictive value of these signatures has not been consistent across different cohorts, rendering its use experimental [99].
In contrast to kidney transplantation, the role of preexisting or de novo anti-HLA donor-specific antibodies in LT recipients is not well established. Although their presence has been associated with allograft injury [100,101] in a single-center cohort, this has not been replicated in other cohorts [102,103]. Interestingly Vionnet et al. demonstrated active "silent" alloimmunity in 36% of the patients with stable graft function using prediction models based on ALT, donor specific-antibodies, and liver stiffness measurements [104].
Serum signatures of immune exhaustion such as high CD45RA T-cells (TEMRA) [105], Eomes+ T cells [98] and PD1+ T Cells [105] hold promise as potential non-invasive biomarkers.
7Invasive biomarkersIn the Barcelona trial [95], adult allograft transcriptional patterns (SOCS1, TFRC, CDHR2, MIF, and PEBP1) offered a PPV of 100%, suggesting a strong association with immune tolerance [99]. This transcriptional signature was predictive independently of the type of immunosuppression used.
CD4 load and the absence of portal inflammation in allograft biopsies performed moderately-weakly in predicting tolerance. To be considered, the inflammatory and regulatory genomic footprints are dynamic [106,107].
In contrast, in pediatric allograft biopsies, the absence of portal inflammation was also strongly associated with immune tolerance. While low leukocyte load, infiltrating monocytes, APC: Lymphocyte pairs, and allograft transcriptional patterns [108] had lower predictive performance, suggesting a different immunological profile between adults and pediatric recipients.
In the case of patients with chronic HCV infection, the allograft expression of an exhausted effector T-cell profile (expressing the inhibitory receptors PD1 and CTLA4) was associated with immune tolerance, suggesting a direct effect of the virus [88]. Interestingly, in a study by Bohne et al. [95], high levels of ferritin, hepcidin-25 (in serum), intra-hepatocytic iron accumulation, and allograft expression of genes associated with iron metabolism predicted tolerance of IS withdrawal [109].
To date, the use of biomarkers for the guidance of IS withdrawal is still experimental, as most have not been prospectively validated, and others have failed to demonstrate external validity. Invasive biomarkers hold promise for greater accuracy [99]. The upcoming trials will aim to collect an ample biorepository to answer this relevant question. Until then, the use of biomarkers should be limited for research purposes (seeFig. 3).
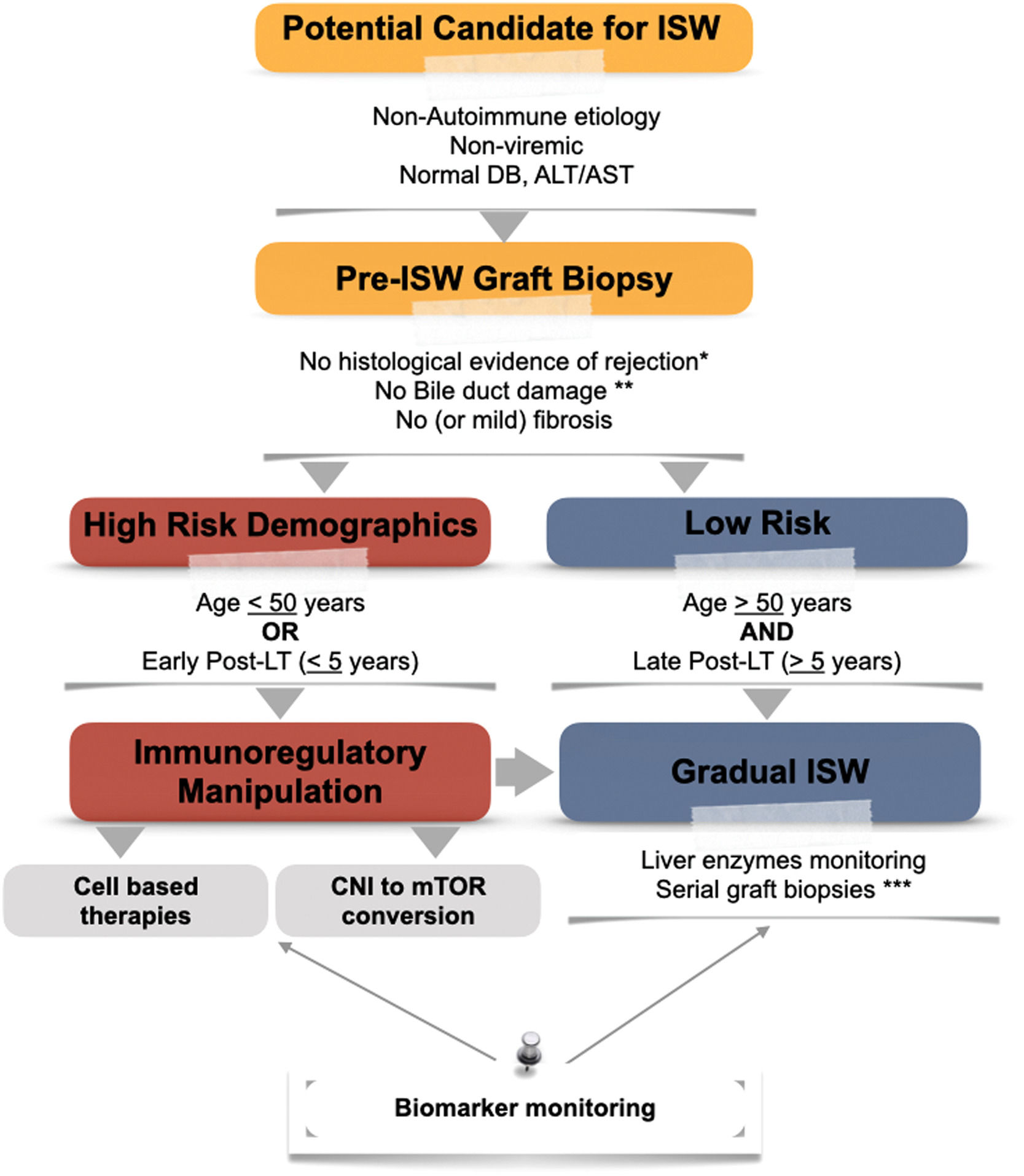
![Proposed algorithm for the approach to immunosuppression withdrawal. CNI, calcineurin inhibitor; DB, direct bilirubin; ISW, immunosuppression withdrawal; LT, liver transplantation; mTOR, mammalian target of rapamycin. * According to Banff Criteria for acute and chronic rejection [81]. **Unless an alternative non-immunological explanation is present. ***Depending on biomarker and liver enzymes monitoring.](https://static.elsevier.es/multimedia/16652681/0000002800000001/v2_202303281200/S1665268122001028/v2_202303281200/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlonC8oCmkTYnB5qUHGrprFXWQfgnWA60C0OegUpCDCS4t4pwCafu8jKRib1MyLogaqKH6qLCLhz6H9lw4XvWkgjvjj8t2QPASjGTMgrVCgCj965GJvEPRzehAu2z6qD4K3BjMnVatf1oK5/aFzvlUV94VaZEn6Q7uECgGSKcnu6j5A7hCKaNMZV4/m1PjNyH4LOMI60POAwQmomoa+DxAYw== "Proposed algorithm for the approach to immunosuppression withdrawal. CNI, calcineurin inhibitor; DB, direct bilirubin; ISW, immunosuppression withdrawal; LT, liver transplantation; mTOR, mammalian target of rapamycin. * According to Banff Criteria for acute and chronic rejection [81]. **Unless an alternative non-immunological explanation is present. ***Depending on biomarker and liver enzymes monitoring.")
Proposed algorithm for the approach to immunosuppression withdrawal. CNI, calcineurin inhibitor; DB, direct bilirubin; ISW, immunosuppression withdrawal; LT, liver transplantation; mTOR, mammalian target of rapamycin. * According to Banff Criteria for acute and chronic rejection [81]. **Unless an alternative non-immunological explanation is present. ***Depending on biomarker and liver enzymes monitoring.
Due to the low percentage of patients who successfully achieved biopsy-proven spontaneous immune tolerance, the interest in pharmacological interventions to achieve it has grown.
Although the aggressive blunting of T-cell responses through anti-thymoglobulin (ATG) has been described in the management of steroid-resistant acute rejection [110,111], its use in tolerance induction (± sirolimus, tacrolimus) has been discouraging [112–114]. A small trial using a combination of ATG and donor-derived mesenchymal stem cells led to a discrete benefit in timing for IS withdrawal. However, phase I trials exploring the use of donor-derived mesenchymal stem cells were proven safe but ineffective for the induction of tolerance in the short and long (85 months) term [115,116]. Trials targeting pediatric LT are still ongoing [117].
Based on the results of experimental models, which demonstrated a decrease in memory CD8+ and an increase in Tregs after the infusion of cDC [118,119] the results of the Pittsburgh trial (the effects of donor-derived CDc infusion before LT in de development of immunotolerance) are eagerly expected [120].
Perhaps the most promising intervention comes from the uncontrolled trial by Todo et al. in Japan [121]. In this study, ten splenectomized patients underwent living donor LT with standard IS plus post-transplantation cyclophosphamide and a single IV infusion of ex-vivo generated donor-derived Treg cells (pre-treated to develop donor-specific immune suppression). After six months of LT (and at least three months of stable graft function), seven patients were successfully withdrawn from IS by month 18th post LT. However, the trial was stopped early (intended to enroll 40 patients) due to the development of mild acute rejection in 3 subjects (all of them with an autoimmune etiology of chronic liver disease), which required longer follow-up for these subjects. Similarly, Sánchez-Fueyo et al. [122] described the safety and tolerability of ex-vivo expanded Treg cells delayed (6 months) infusion after LT. Although the immunological characterization suggested a potential benefit in donor-specific responses, IS withdrawal was unsuccessful. Based on these studies, the results of the ARTEMIS (NCT02474199), LITTMUS-MGH (NCT03577431), and Nanjing University (NCT01624077) trials are eagerly awaited [123]. Potential alternatives to ex-vivo expanded Tregs are using CAR-T cells [124,125], IL-2/Il-10 mediated Treg in-vivo induction [126,127], or genome editing via CRISPR-Cas9 [128], all of which are still in the experimental phase.
9ConclusionThe therapeutic applications of recent discoveries in immunotolerance mechanisms are shifting gears toward a personalized medicine approach to IS withdrawal. Further research will shed light on risk prediction tools for guiding the patient selection and adequate timing for IS withdrawal. For patients in whom IS withdrawal was unsuccessful, or those who require IS withdrawal despite a high-risk profile (due to IS toxicity and oncologic complications), the development of pharmacological interventions to induce immune tolerance is much needed.
Author contributionsConceived and designed the study: JPE, JVJ, and IGJ; performed Literature Review: JPE, JVJ, EFR, MSR, JRM, LSB, ECM, MN and IGJ; wrote the paper: JPE, JVJ, EFR, MSR, JRM, LSB, ECM, MN and IGJ. All authors have approved the final draft of the manuscript. The corresponding author attests that all listed authors meet authorship criteria.
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.



