



Today the knowledge of genotype-phenotype correlation in cystic fibrosis is enriched by the growing discoveries of new mutations of the CFTR gene. Although the combination of two severe mutations usually leads to the classic disease (pulmonary and pancreatic insufficiency, sterility, nasal polyposis), the presence of a complex genotype characterized by severe and milder mutations or polymorphism can cause a hidden disease, which is often asymptomatic at early ages. We report on a case of a 15 years old boy, in whom the only clinical signs of CF were chronic hypertransaminasemia and hyperbilirubinemia, and in whom it was demonstrated the presence of the mutations F508del associated with TG11-9T-470M in one allele and TG12-5T-470V in the other allele. Although a clear genotype-phenotype correlation for liver disease is still missing for CF patients, it is possible to state that this isolated clinical presentation could represent an unusual phenotype of CF, related to a complex genotype characterized by a severe mutation and one (or more) polymorphism.
Prevalence of liver disease in patients affected by cystic fibrosis (CF) is about 30-40% and it is the third cause of death in these patients.1,2 Hypertransaminasemia and hyperbilirubinemia are the main biochemical manifestations, and usually occurs in 50% of patients,3 while the most frequent hepatic disorders are hepatic steatotis (23-67%), focal biliary cirrhosis (11-72%), multilobar cirrhosis (5-10%), neonatal cholestasis and liver failure.1,4
The exact pathogenesis of liver disease in CF seems to be related to the alteration of the cystic fibrosis transmembrane regulator (CFTR) channel, located on the apical side of the biliary ducts, resulting in a dehydrated and viscous bile secretion, which leads to obstruction of small biliary ducts. According to this etiopathogenetic hypothesis, cholestasis could activate an inflammatory cascade with production of fibrogenic molecules.4 Moreover liver damage could be modulated both by genes modifiers and environmental factors.5 A clear genotypephenotype correlation for the occurrence liver disorder in cystic fibrosis has not been demonstrated, even if liver disease is more frequent in patients carrying severe genotypes.4
In the last years, the discovery of new genetic mutations has allowed to explain many uncommon forms of CF6,7 and in many of these cases, genotypephenotype correlation is resulted difficult to clarify, especially when a CF-causing mutation is combined with one or more milder mutations or polymorphisms. Furthermore, the phenotype of patients affected by CF can be influenced by environmental factors (infections or pollution) or other gene modifiers, thus making almost unpredictable the course of the disease in subjects with rare genotypes.8
In this setting, a new field of research is represented by the possible phenotypic expressions of patients carrying poly-5T variants in association with disease causing mutations, such as F508del. 5T is a widely expressed allelic polymorphism, approximately presented by 1 in 10 individuals, and when it is combined with another CF-causing mutation, it can lead either to CF phenotype or to a healthy condition,9 in particular if associated with a longer (12 or 13) TG tract. The most common clinical feature presented by patients with this genotype is the congenital absence of vas deferens (CABVD), pancreatic sufficiency, and normal lung function.9,10
Although quite common in the classic form of the disease,4 apart from CBAVD, to date, liver disease has never been reported as manifestation of a F508del/ 12TG-5T-470MV genotype. We herein describe a young adult patient admitted to our unit because of a history of jaundice and hypertransaminasemia, and in whom a diagnosis of CF caused by this genotype was performed.
Case ReportThe patient, a 15-year old boy was recovered at our unit, tertiary referral centre for Child Pneumoallergology and Cystic Fibrosis, with a two years history of chronic hypertransaminasemia and jaundice. The boy was born at-term with the weight of 3.4 kg. Family and personal history were negative and the patient did not report any exposure to toxic substances or drugs in the last two years. At physical examination, weight and height were respectively 42.7 kg (3rd percentile) and 160 cm (10-25th percentile) and, scleral jaundice was the only clinical sign. Blood tests showed aspartate aminotransferase (AST) value of 66 U/I (n.v. < 40 U/I) and alanine aminotransferase (ALT) of 200 U/I (n.v. < 40 U/I), increased alkaline phosphatase (646 U/I, n.v. = 300-400 U/I), and hyperbilirubinemia (total bilirubin 3.48 mg/dL, indirect bilirubin 1.84 mg/dL). All the other blood tests, including full blood count, glycaemia, lipid profile, electrolytes, protein electrophoresis, coagulation tests, renal and pancreatic function tests, blood iron subset and immunoglobulins, were within normal range. The results of bacterial and viral assessment were negative, and so the tests for Wilson disease, autoimmune hepatitis, non-alcoholic fatty liver disease, celiac disease and hereditary hemochromatosis.
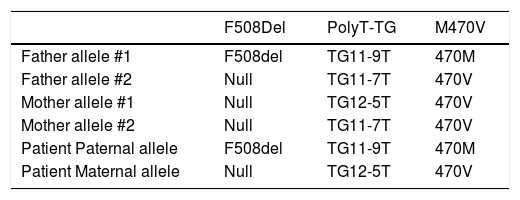
An abdominal ultrasound showed a slight increase in liver size and hyperechogenity of the parenchyma. A liver biopsy was performed, and there was no evidence of anomalies at the histological examination. A sweat chloride test was performed, with a result of 72 mEq/L (n.v. < 40 mEq/l). Two other tests were performed to confirm this result. The second showed a borderline result (47 mEq/L) and the third a positive result (60 mEq/L). For this reason, a genetic first level investigation for Cystic Fibrosis performed by reverse dot blot commercial kits (INNO-LIPA CFTR19, INNO-LIPA CFTR17+TnUpdate and INNO-LIPA Italian-Regional, Innogenetics, Ghent, Belgium) showed the presence of heterozygous mutation F508del and the presence of the 5T polymorphism. A second level investigation, performed by automated DNA sequencing using ABI Prism 3100 (Applied Biosystems, Warrington, UK), confirmed the presence of the haplotype TG12-5T plus the 470V variant in one allele and F508del and TG11-9T plus the 470M variant in the other. The same genetic analyses were performed in the parents and results are showed in table 1. The presence of this genotype led to the diagnosis of CF.
Genotype of the patients and of his parents.
| F508Del | PolyT-TG | M470V | |
|---|---|---|---|
| Father allele #1 | F508del | TG11-9T | 470M |
| Father allele #2 | Null | TG11-7T | 470V |
| Mother allele #1 | Null | TG12-5T | 470V |
| Mother allele #2 | Null | TG11-7T | 470V |
| Patient Paternal allele | F508del | TG11-9T | 470M |
| Patient Maternal allele | Null | TG12-5T | 470V |
The bacteriological analysis of sputum and a spirometry were performed, both with normal results. Chest X rays evaluation was normal. Pancreatic function was normal, as revealed by a fecal elastasys value of 400 µg/g, and normal levels of amylase and lipase.
Therapy with ursodeoxycholic acid at a dose of 15 mg/kg/day was initiated, with a slighter reduction of transaminases value (AST 60 U/L; ALT 110 U/L), but persistence of high bilirubin level (3.19 mg/dL). After six months of therapy, transaminases values decreased to normal levels (AST 30 U/L; ALT 35 U/L), and bilirubin level was further decreased 2.22 mg/dL. The presence of CBAVD was initially suggested by palpable scrotal vas on physical examination and by the results of a trans-rectal ultrasonography and subsequently confirmed by MRI examination of the seminal vesicle (SV) and intra-abdominal segment of vas deferens. The parents of the patient refused to sign the consent to perform a sperm count.
DiscussionBefore the introduction of mandatory neonatal screening (in Sicily from 1999), CF was diagnosed at early age only if patients presented severe symptoms such as neonatal meconium ileus, recurrent Pseudomonas aeruginosa lung infections and pancreatic insufficiency.11 At present, it is not rare, for patient born before 1999 or negative to the screening, or affected by atypical or milder form of cystic fibrosis, to be diagnosed in adolescence or adulthood, by the genetic evaluation.7,8
On this regard, complex genotypes are becoming a common finding in patients with these milder presentations, which usually consist in involvement of only one system (i.e. isolated pancreatic insufficiency, or CBAVD), in absence of severe symptoms. The genotypes of these patients can be characterized by one severe mutation plus one milder mutation, two or more milder mutation, one severe mutation plus one or more polymorphisms etc. Isolated liver disease is very rare and can represent an example of the role of genetic polymorphisms, which can express with a normal lung and pancreatic function, but with other organs dysfunction, such as CBAVD.
Polymorphisms are common variant of a gene, usually found in > 1% of the population. Particular polymorphisms, such as 5T, or TG, M470V are commonly found in the healthy population, but if combined with severe CF-causing mutations, they can lead to unpredictable form of disease.12
The 5T allele in intron 8 (IVS8) is a mild variant of CFTR, resulting in mRNA lacking exon 9 and for this reason, 5T is thought to decrease the efficiency of intron 8 splicing.9 The association F508del and 5T has been related to a less functional or even insufficient CFTR protein, leading to atypical CF or CBAVD in 60% of the cases.13–15 Other common variants of the IVS8, both considered polymorphic, are 7T and 9T, which differently from 5T do not cause atypical CF or CBAVD, even when associated to F508del or other severe mutations.9
When a 5T mutation is present, the percentage of functioning protein is strictly related to the repetition of a TG tracts which lies in proximity to poly T and consists of a short string of TG repeats that commonly number 11, 12, or 13. A longer TG tract (12 or 13) in conjunction with a shorter poly T tract (5T) has the strongest adverse effect on proper intron 8 splicing and it is associated with higher values at IRT test.16 Our patient presented a TG12-5T mutation, which has been reported to cause CF symptoms or CBAVD in 78% of affected patients.9
A part from the higher risk of CBAVD, the presence of a IVS8 mutation in combination with a severe mutation on the other allele has been more rarely associated to severe lung disease,17 and to recurrent acute pancreatitis.18
The expressivity of poly-T and TG tract variant can be further complicated by the presence of 470M or 470V haplotype, which are both considered normal. M470V (1540A/G in exon 10) locus is polymorphic at amino acid level, and causes a different maturation and intrinsic chloride channel activity: 470M CFTR protein matures more slowly and has 1.7-fold increased channel activity compared to 470V protein.8 Although 470M and 470V alleles differ functionally, there have been no reports that they alone could be associated with CF, but is much likely that they have synergistic effect with other sequence variants such as poly-T or TG tract. As reported by Noone and coll., the 5T polythymidine tract sequence on specific haplotype backgrounds (TG12-470M) may cause a low level of full-length functional CFTR protein and CF-like lung disease.19 In a more recent paper it was showed that patients with 470V genotype had a 3.4-fold decreased risk of severe pulmonary disease.20
Differently, the main symptom presented by our patient was an isolated liver involvement consisting in persistent hyperbilirubinemia and hypertransaminasemia, never reported as isolated feature in patients carrying these mutations.
In our case, the presence of the severe mutation F508del associated with TG11-9T-470M on one allele and a pattern of milder polymorphism TG12-5T-470V on the other chromosome, represents a never reported genotypic profile, in which lung and pancreas do not seem to be affected, CBAVD is probably present and liver involvement is mild with no specific pathological findings at liver biopsy.
ConclusionsAlthough a clear genotype-phenotype correlation for liver disease is still missing for CF patients, it is possible to state that this isolated clinical presentation could represent an unusual phenotype of CF, related to a complex genotype characterized by a severe mutation and one (or more) polymorphism.
Authors’ ContributionADP wrote the entire manuscript, ERP and SS revised the literature, NR and CF followed up the patient, SL revised the entire manuscript.
Andrea D. Praticò is responsible for the integrity of the work as a whole.
Conflict of InterestsThe authors have nothing to disclose.



