





Background: Non-alcoholic fatty liver disease (NAFLD) is a prevalent condition associated with obesity and insulin resistance (IR). Leptin plays a key role in the control of energy balance, and insulin sensitivity. In this study, we aimed to examine whether serum leptin levels correlate with insulin resistance, oxidative stress parameters and the severity of histological changes in NAFLD. Methods: Fifty-two patients (M/F: 28/24) with no alcohol intake and biopsy-proven diagnosis of NAFLD were studied. Serum leptin levels were measured by radioimmunoassay. HOMA (homeostasis model assessment) IR index was calculated. Comparisons between the patients with NAFLD and non-alcoholic steatohepatitis (NASH) were performed using the Student’s t test. Multivariate regression analysis and the area under the receiver operating characteristic (ROC) curve were used to identify the independent predictors for NASH. Results: We found no association between serum leptin, fasting insulin levels, and oxidative stress parameters. ROC curve and multiple regression analysis revealed no association between the severity of histological changes and serum leptin levels. During six months followed-up period only NASH group with elevated leptin levels had significant reductions of ALT and AST values (p = 0.03, and 0.005, respectively). Conclusion: Our findings show a preventive effect of leptin against progressive liver injury in NAFLD.
Non-alcoholic fatty liver disease (NAFLD) affects 10 to 24 percent of the general population.1 The clinical spectrum of NAFLD ranges from simple steatosis to steatohepatitis (NASH) and even to cirrhosis.2-6 NASH frequently occurs in the setting of obesity and insulin resistance.7-9 Leptin is mainly expressed by adipose tissue. Leptin decreases food intake, and increases energy expenditure.10-14 The principal role of leptin may be to limit the accumulation of fat in non-adipose tissue, and reduce lipotoxicity.15 By increasing the supply of free fatty acids (FFA) to the liver, leptin induces dephosphorylation of insulin receptor substrate 1 to prevent fat accumulation in this organ.16 Hepatic stellate cells (HCS’s) are the primary source of extracellular matrix proteins, and accordingly fibrosis. Previous observations showed that activated, HSCs express leptin. These findings pointed to the implication of leptin in the fibrogenesis, and the disease progression.17,18 However these theoretical concerns were challenged in clinical studies.19,20 On the other hand, by increasing reactive oxygen species (ROS) leptin may induce Kupffer cells to produce inflammatory cytokines such as tumor necrosis factor-α (TNF-α) which interacts with stellate cells and augments collagen gene expression.21-23 In this study, we aimed to examine whether serum leptin levels correlate with insulin resistance, oxidative stress parameters and the severity of histological changes in NAFLD.
MethodsPatientsFifty-two non-drinking patients with biopsy proven diagnosis of NAFLD were enrolled into the study in two university hospital clinics. Patients were referred for the assessment of abnormal liver function tests or hepatic steatosis detected by ultrasonography. Most of these patients were referred from the internal medicine outpatient clinic and endocrinology department of the same hospitals. Viral hepatitis, autoimmune hepatitis, primary biliary cirrhosis, α1-antitrypsin deficiency, Wilson’s disease, hemochromatosis, and sclerosing cholangitis were excluded with relevant tests. Informed consent was obtained from each of the participating subject. Steatosis or hepatomegaly in US was considered enough to perform a liver biopsy after exclusion criteria in patients with elevated ALT. Patients were followed up for six months with a dietary advice.
Pathological examinationLiver tissues were stained with hematoxylin-eosin and reticulum. Histological examination was done by an experienced pathologist (G.O.) who was blinded to clinical information using the scoring system described by Brunt et al.24 We defined simple steatosis as steatosis without fibrosis and inflammation, and NASH as steatosis with inflammation, ballooning degeneration of hepatocytes, and zone 3 perisinusoidal fibrosis. Steatosis was graded from 1 to 3 (grade 1 up to 33%; grade 2 33-66%; grade3 > 66%). The combination of hepatocellular steatosis, ballooning, acinar or portal inflammation, and zone 3 pericellular fibrosis was used for necroinflammatory grading from 0 to 3. The severity of hepatic fibrosis (stage) was graded with a 4-point scale: stage 1, zone 3 perisinusoidal/ pericellular fibrosis; stage 2, zone 3 perisinusoidal/ pericellular and periportal fibrosis; stage 3, zone 3 perisinusoidal/ pericellular portal and bridging fibrosis; stage 4, cirrhosis.
Laboratory evaluationsOn the morning of liver biopsy venous blood samples were drawn after an overnight 12-hour fasting period. The serum levels of ALT, AST, alkaline phosphatase (ALP), gamma-glutamyltranspeptidase (GGT), bilirubin, total cholesterol, triglyceride, fasting insulin, glucose and leptin levels were measured. Total cholesterol, triglyceride were measured by enzymatic methods and glucose was determined by hexokinase method using Abbott C8000 (Abbott, USA) automatic analyzer. Insulin was measured with Immulite 2000 analyzer (DPC, USA) by Chemiluminescent immunometric assay.
Serum Leptin was measured using a human IRMA kit (DSL, Texas, USA). The IRMA is a non-competitive assay in which leptin is sandwiched between two antibodies. Samples were incubated overnight in antibody-coated tubes with the secondary radio-labeled antibody.
The sensitivity of the assay was 0.10 ng/mL, and the intra-and interassay coefficients of variation (CVs) were 2.6-4.9% and 3.7-6.6%, respectively. The standards ranged from 0.25 to 120.0 ng/mL.
We calculated insulin resistance using the homeostasis model assessment (HOMA-IR) formula [HOMA-IR = fasting glucose (mmol/L) x fasting insulin (μU/mL)/22.5].
Oxidative stress parameters
Malondialdehyde (MDA), and superoxide dismutase (SOD) levels were measured in serum as well as in liver tissue homogenates. TNF-alpha receptor (TNF-sRp55) and nitric oxide (NO) levels were measured in sera. MDA as an end product of fatty acid peroxidation was measured by the thiobarbituric acid reactivity assay as previously described. 25 Serum and tissue SOD activities were determined by inhibition of nitroblue tetrazolium reduction by Xanthine/Xanthin oxidase used as a superoxide generator. 26 Nitric oxide level was determined with Griess reaction. A commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D systems, Quantikine; Wiesbaden - Nordenstadt, Germany) was used. Serum TNF-sRp55 concentrations were measured using ELISA (Hbt, HyCult Biotechnology; Uden, the Netherlands).
Statistical analysisComparison between the patients with simple steatosis and patients NASH were made with the Student’s ttest or Mann-Whitney test. Chi square test was used for nominal categorical variables, and the Spearman rank correlation was used to assess the association between continuous variables. Multiple regression analyses, binary logistic regression and linear regression analysis were performed to identify the independent predictors for the degree of steatosis, inflammation, and fibrosis. Area under the receiver operating characteristic (ROC) curve was used to demonstrate the diagnostic ability of HOMA-IR, and serum leptin levels to distinguish between simple steatosis and NASH. Results are expressed as the mean ± SD. All analyses were performed with SPSS software for Windows, version 10. P value below 0.05 was considered statistically significant.
ResultsCharacteristics of the patientsFifty-two non alcohol drinking patients (male/female: 28/24; mean age ± SD: 48.34 ± 9.8) with NAFLD were included in the study. There was no difference in reference to age between males (mean age ± SD: 44.25 ± 8.75) and females (mean age ± SD: 47.12 ± 9.07). Among patients, 16/35 (46%) patients with NASH and 7/17 (41%) subjects with simple steatosis were type 2 diabetic.
Serum leptin levels were significantly higher in females (r = 0.48, p = 0.0001), and positively correlated with body mass index (BMI) (r = 0.43, p = 0.002). However females had significantly higher BMI than the males (31.92 ± 5.36 and 29.24 ± 4.14, respectively, p = 0.04). Serum leptin was, independently, associated with BMI (p = 0.002) but not with gender. Serum leptin levels did not correlate with serum ALT (r = -0.03, p = 0.83), or AST(r = 0.15, p = 0.28) values. To assess whether serum leptin had an effect on insulin resistance, we closely examined the correlation between leptin and carbohydrate metabolism. We found no association between serum leptin levels and the fasting insulin (r = -0.13, p = 0.5), serum glucose (r = 0.2, p = 0.13), and HOMA-IR (r = -0.07, p = 0.6).
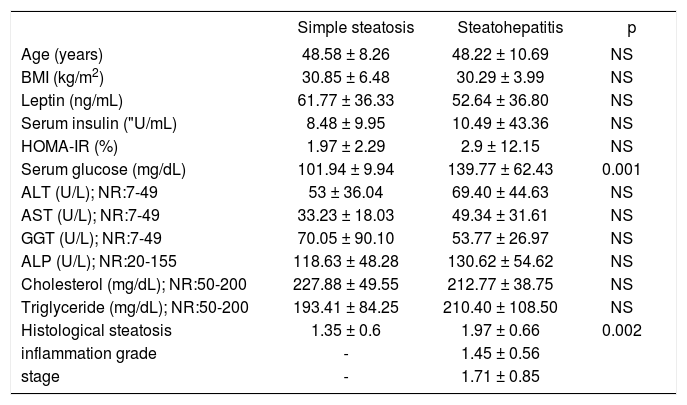
Comparative analysis between subjects with simple steatosis and NASHThe mean leptin levels tended to be higher in the group with simple steatosis than the NASH group but the difference between the group was not significant. Demographic, laboratory and histopathological data of patients with simple steatosis and NASH are summarized in table I
Demographic, laboratory and histopathological data of the patients with non-alcoholic simple steatosis, versus steatohepatitis. BMI: Body mass index, GGT: Gamma glutamyl transpeptidase, ALP: alkaline phosphatase, NR: normal range.
| Simple steatosis | Steatohepatitis | p | |
|---|---|---|---|
| Age (years) | 48.58 ± 8.26 | 48.22 ± 10.69 | NS |
| BMI (kg/m2) | 30.85 ± 6.48 | 30.29 ± 3.99 | NS |
| Leptin (ng/mL) | 61.77 ± 36.33 | 52.64 ± 36.80 | NS |
| Serum insulin ("U/mL) | 8.48 ± 9.95 | 10.49 ± 43.36 | NS |
| HOMA-IR (%) | 1.97 ± 2.29 | 2.9 ± 12.15 | NS |
| Serum glucose (mg/dL) | 101.94 ± 9.94 | 139.77 ± 62.43 | 0.001 |
| ALT (U/L); NR:7-49 | 53 ± 36.04 | 69.40 ± 44.63 | NS |
| AST (U/L); NR:7-49 | 33.23 ± 18.03 | 49.34 ± 31.61 | NS |
| GGT (U/L); NR:7-49 | 70.05 ± 90.10 | 53.77 ± 26.97 | NS |
| ALP (U/L); NR:20-155 | 118.63 ± 48.28 | 130.62 ± 54.62 | NS |
| Cholesterol (mg/dL); NR:50-200 | 227.88 ± 49.55 | 212.77 ± 38.75 | NS |
| Triglyceride (mg/dL); NR:50-200 | 193.41 ± 84.25 | 210.40 ± 108.50 | NS |
| Histological steatosis | 1.35 ± 0.6 | 1.97 ± 0.66 | 0.002 |
| inflammation grade | - | 1.45 ± 0.56 | |
| stage | - | 1.71 ± 0.85 |
NS = statistically not significant; NR= normal range.
In bivariate analysis there was no significant association between serum leptin levels and the degree of hepatic steatosis (r = 0.04, p = 0.75), hepatic inflammation (r = -0.01, p = 0.92), or hepatic fibrosis (r = 0.04, p = 0.47) in NASH.
Predictors for the histological severity of NAFLDIn a multivariate comparison between subjects with NASH, and subjects with simple steatosis we entered HOMA-IR, serum leptin, serum insulin, and serum glucose into the model. Only the increased serum glucose level was a risk factor for the histological disease severity and leptin had preventive effect. The overall accuracy of this analysis was 77.5% (Table II)
In a multivariate comparison between subjects with NASH, and subjects with simple steatosis only the increased serum glucose level was a risk factor for the histological disease severity. Leptin had preventive effect. The overall accuracy of this analysis was 77.5%. HOMA-IR: homeostasis model assessment - Insulin resistance.
| Characteristics | Odds ratio (OR) | 95% CI (Confidence interval) | p |
|---|---|---|---|
| Leptin (ng/mL) | 0.94 | 0.90-0.99 | 0.02 |
| Serum insulin (aU/mL) | 0.41 | 0.15-1.07 | 0.07 |
| HOMA-IR (%) | 0.97 | 0.81-1.6 | 0.44 |
| Serum glucose (mg/dL) | 1.09 | 1.0-1.19 | 0.03 |
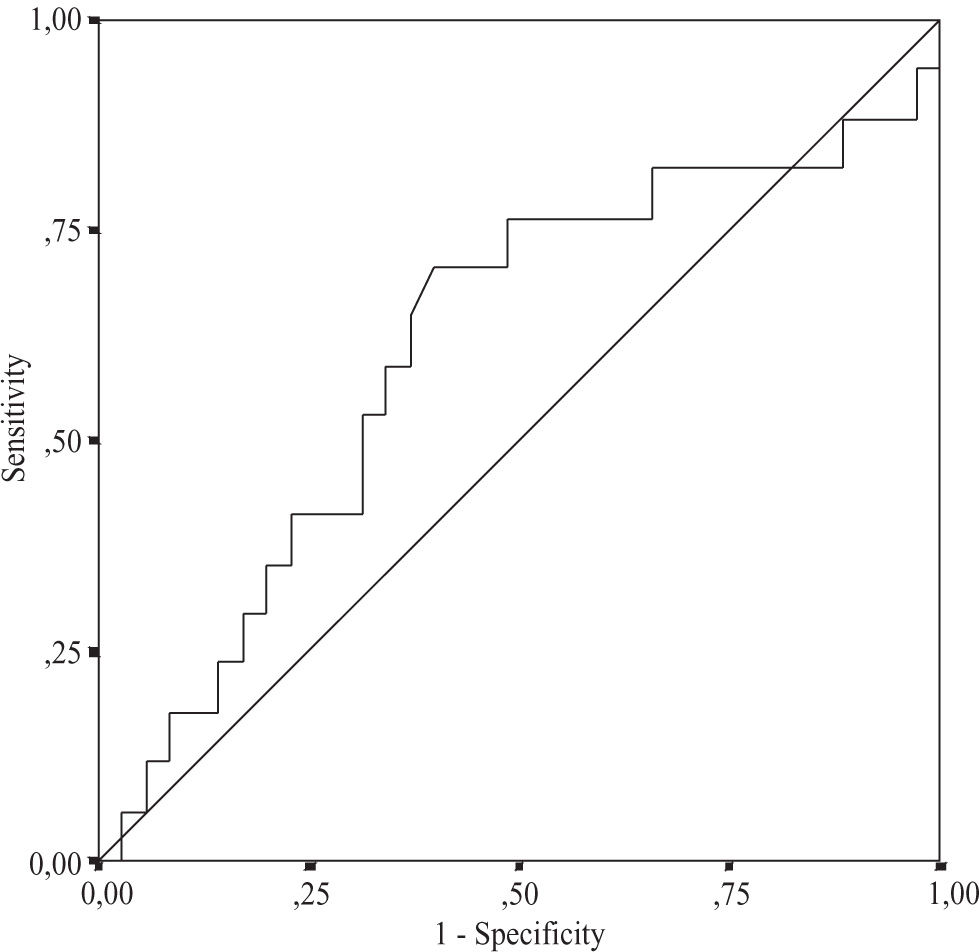
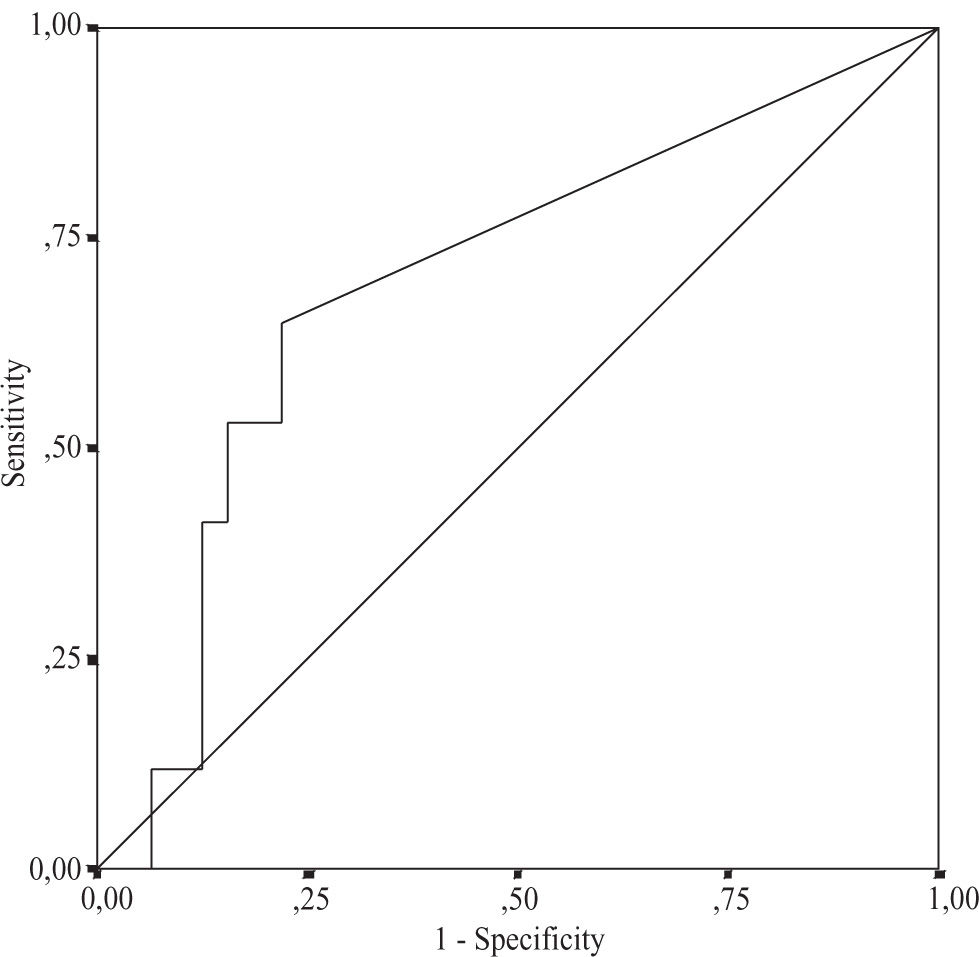
Using multiple regression analysis the independent predictors for the grade of steatosis, inflammation, and fibrosis were investigated. The relationship between the severity of histological changes in NASH and serum leptin, serum glucose, serum fasting insulin levels, and HOMAIR was analyzed. None of these variables were independent predictors for disease severity (Table III) Using serum leptin levels and HOMA-IR the area under the ROC curve for distinguishing between NASH and simple steatosis was 0.60, 95% CI 0.46-0.74, and 0.69, 95% CI 0.53-0.85, respectively (Figure 1andFigure 2). The serum HOMA-IR cut off value for the prediction of NASH was £ 0.1%; at this threshold the sensitivity was 78.1% and the specificity 64.7%. The sensitivity for leptin at the threshold 50 ng/mL was 60% and the specificity 70.6%. Both curves didn’t show any respective sensitivity and specificity for the differentiation between simple steatosis and NASH.
Multiple regression did not show any correlation between laboratory parameters studided and histological severity in patients with steatohepatitis. HOMA-IR: homeostasis model assessment - Insulin resistance.
| Characteristics | Steatosis OR | Inflammation 95%CI | grade p | Stage OR | 95%CI | p | OR | 95%CI | p |
|---|---|---|---|---|---|---|---|---|---|
| Leptin (ng/mL) | 1.03 | 0.99-1.07 | 0.11 | 0.99 | 0.98-1.01 | 0.88 | 0.97 | 0.93-1.02 | 0.33 |
| Serum insulin ("U/mL) | 0.96 | 0.35-2.63 | 0.94 | 0.80 | 0.58-1.10 | 0.17 | 0.41 | 0.05-3.26 | 0.40 |
| HOMA-IR (%) | 0.92 | 0.01-89.62 | 0.97 | 0.40 | 0.10-1.50 | 0.17 | 27.71 | 0.01-7.52 | 0.41 |
| Serum glucose(mg/dL) | 0.98 | 0.94-1.02 | 0.28 | 1.01 | 0.99-1.02 | 0.07 | 0.99 | 0.97-1.0 | 0.36 |
OR: Odds ratio; CI: confidence intervals
 (area under the ROC curve 0.6; 95 % CI: 0.460.74).")
 (area under the ROC curve 0.69; 95 % CI: 0.53-0.85).")
Serum leptin levels were neither correlated with serum NO, SOD and MDA levels (r = 0.09, p = 0.49; r = -0.35, p = 0.06; r = -0.12, p = 0.53, respectively) nor with tissue SOD and MDA levels (r = -0.22, p = 0.23, and r = -0.34, p = 0.85, respectively). The increases in plasma TNF-sRp55 levels correlated inversely with serum leptin levels (r = - 0.03, p = 0.04).
Follow-up periodAfter six months, in NASH patients with more than the mean leptin levels had significant reductions of ALT and AST values from 82.25 ± 11.26 to 49.83 ± 10.46, p = 0.003, and from 71.58 ± 11.46 to 41.5 ± 7.32, p = 0.005, respectively. In this group, patients with less than the mean leptin levels had no significant reductions in ALT and AST values (p = 0.06, and 0.45, respectively). The changes in relevant biochemical parameters were also not significant in patients with simple steatosis.
DiscussionThere are conflicting reports about the exact role of leptin in the pathogenesis of NAFLD. Obesity and insulin resistance are the most consistently associated causal factors in NAFLD.7-9 Recent experiments showed that leptin promotes insulin resistance by inducing the dephosphorylation of insulin-receptor-substrate 1.16,27 Some studies have shown that leptin administration might actually improve the insulin resistance in patients with lipodystrophy and in mice with congenital lipodystrophy. 28,29 We did not find any significant association between serum leptin levels and insulin resistance. Linear regression analysis showed that serum leptin level was not an independent predictor for an increased HOMA-IR.
Activated HCS’s are the main course of fibrogenic actions. Isolated HSC’s have been shown to produce leptin. 17,30-32 This finding indicates that leptin might play a pivotal role in profibrogenic responses in the liver. Whether leptin levels are altered in NAFLD is controversial. There are only few studies done in humans with NAFLD. The published reports have yielded conflicting results regarding the role of leptin in disease progression. Some studies demonstrated increased leptin levels in NASH, while others found no correlation between serum leptin levels and the development of NASH.33,34 Most of the studies are based on animal models. In ob/ob mice lacking circulating leptin, impaired wound healing and surgical scar formation was documented.35 Lean animals but not ob/ob littermates had significant fibrosis.36,37 Increased fibrosis and greater expression of procollagen type I were observed when leptin was injected into rats receiving carbon tetrachloride or thioacetoamide to produce an acute or chronic liver damage, respectively.30
Histopathology is most relevant in grading the severity of NAFLD. Therefore we focused on comparative analysis between subjects with simple steatosis and NASH. Mean leptin levels were higher in the group with simple steatosis compared to the NASH group. In bivariate analysis there was no significant association between serum leptin levels and the degree of hepatic steatosis, hepatic inflammation, or hepatic fibrosis in NASH. Using serum leptin levels, the area under the ROC curve for distinguishing between NASH and NAFL did not show any respective sensitivity and specificity for leptin levels. Moreover in multivariate comparison serum leptin had low preventive effect against steatohepatitis. Our data excludes the possibility that leptin leads to disease progression.
Some studies have shown that leptin contributes to oxidative stress and necroinflammation.38,21 Leptin-deficient mice had less necroinflammatory activity in models of T-cell mediated hepatitis which was induced by injection of concavalin A.39 In another study macrophages expressed the functional long form of the leptin receptor, which indicates that this hormone can attenuate inflammatory reaction.17
To determine the effect of leptin in necroinflammatory activity we investigated the correlation between serum leptin levels and the oxidative parameters. In our study leptin levels didn’t correlate significantly with oxidative stress parameters measured in sera as well as in tissue homogenates. This data also argues against a role for leptin in NAFLD progression.
Higher ALT levels and increase in serum TNF-α levels were observed when carbon tetrachloride intoxication was accompanied by injection of leptin.30 Conversely, leptin deficiency was associated with increased hepatotoxicity and mortality following endotoxin administration, 40 an effect mediated by increased sensitivity to TNF-a.41 These indicate the protective effect of leptin against hepatic injury.
Our findings support this latter suggestion. The regression analysis showed that leptin has protective effect against the development of NASH. Leptin levels correlated inversely with TNF-sRp55 levels. Moreover, during the follow-up period patients with NASH and increased leptin levels achieved significant reductions in ALT and AST values.
In conclusion, there was no significant association between serum leptin levels, fasting serum insulin levels, and HOMA-IR. This argues against a potential role played by leptin in insulin resistance development.
Our findings did not show any correlation between serum leptin levels and oxidative stress parameters and overall histological severity. Furthermore in NASH patients with elevated leptin levels, improvement in ALT and AST values with dietary advice was significantly higher. This indicates that leptin may have preventive effect against progression of hepatic injury.



