






Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) includes liver disease processes from simple fatty liver to nonalcoholic steatohepatitis, which may progress to liver fibrosis, cirrhosis, and even hepatocellular carcinoma (HCC). As the incidence of HCC derived from viral hepatitis decreases, MASLD has emerged as a significant health threat, driven by lifestyle changes and rising obesity rates among patients. The pathogenesis of MASLD is complex, involving factors such as insulin resistance, gut microbiota imbalance, and genetic and epigenetic factors. In recent years, the role of mitochondrial dysfunction in MASLD has gained significant attention, involving β-oxidation imbalance, oxidative stress increase, mitophagy defects, and mitochondrial DNA (mtDNA) mutations. This article reviews the pathophysiological mechanisms of mitochondrial dysfunction in MASLD, diagnostic methods, and potential therapeutic strategies. By synthesizing current research findings, the review aims to highlight the critical role of mitochondrial dysfunction as a target for future diagnostic and therapeutic interventions. This focus could pave the way for innovative clinical strategies, ultimately improving treatment options and patient prognosis in MASLD.
Recently, "Non-Alcoholic Fatty Liver Disease" (NAFLD) has been renamed to “Metabolic Dysfunction-Associated Steatotic Liver Disease” (MASLD). This new term, "Metabolic Associated Steatotic Liver Disease" (MASLD), underscores the critical role of metabolic factors in the diseaseʼs progression, such as obesity, diabetes, and hyperlipidemia [1]. With the increasing prevalence of metabolic syndrome, MASLD has become a common liver condition [2]. As of 2019, MASLD affected 38 % of the global population [3,4]. Chris Estes and his team utilized mathematical models to study the status of MASLD in countries including China, the United States, France, and Japan from 2016 to 2030. Their findings indicated that China had the highest number of MASLD cases worldwide, with the patient population expected to grow from 246.33 million in 2016 to 314.58 million by 2030, an increase of approximately 29.1 % [5]. MASLD exhibits a strong association with hepatocellular carcinoma (HCC), with its microenvironment fostering HCC cell proliferation and migration [6]. In recent years, the incidence of MASLD-driven HCC has been increasing [7]. MASLD has become a global health problem that brought huge medical and economic burden [8].
Increasing evidence indicated that mitochondrial dysfunction is associated with the progression of MASLD. During oxidative stress, the activity of the mitochondrial electron transport chain (ETC) is inhibited, leading to increased generation of reactive oxygen species (ROS), which plays an important role in the pathogenesis of MASLD [9]. Blocked mitochondrial β-oxidation in the liver leads to the accumulation of fatty acid, the generation of lipotoxicity, and the activation of pro-inflammatory signaling pathways. In addition, excessive reactive oxygen species can lead to liver cell damage and fibrosis, which are generated by oxidative stress. Furthermore, mitophagy defects hinder the timely clearance of defective mitochondria and aggravate cell damage. Mutations and damage of mitochondrial DNA further impair mitochondrial function and are one of the causes of energy metabolism disorders in MASLD [10–14].
We review the mechanisms of MASLD development and the structure and function of mitochondria, highlighting mitochondrial dysfunction as a key risk factor for MASLD. Finally, we discussed the significance of targeting mitochondrial dysfunction for diagnosis and treatment, aiming to improve therapy and patient outcomes (Fig. 1).

Schematic representation of the mechanisms of MASLD and potential therapeutic interventions. MASLD progression is linked to mitochondrial ROS, dysregulation of β-oxidation in PDM, mtDNA mutations, and mitophagy disorders. These mitochondrial dysfunctions contribute to disease progression but can be targeted by various interventions. Non-pharmacological approaches, such as physical activity, and pharmacological strategies, including drugs, chemical agonists/inhibitors, and natural extracts, can promote MASLD remission by addressing mitochondrial dysfunction. MASLD, metabolic dysfunction-associated steatotic liver disease.
In 1998, Day and James proposed the “two-hit” hypothesis to explain how simple steatosis progresses to nonalcoholic steatohepatitis (NASH) in MASLD. The first hit is the accumulation of fat in the liver, which is driven by lifestyle, dietary habits, obesity, and insulin resistance. The second hit involves inflammation and oxidative stress, leading to hepatocyte damage, inflammation, and fibrosis, which promote the progression of MASLD [15]. Although significant hepatic lipid accumulation is observed in the genetic obesity ob/ob mouse model of leptin deficiency, a second hit is required to induce inflammation and fibrosis [16]. As research on MASLD has advanced, it has become clear that the "two-hit" model is too simplistic to fully capture the complexity of the disease. The current trend in MASLD research favors the “multiple-hit”, which offers a more comprehensive explanation of the disease's development by suggesting that various factors—including insulin resistance, immune dysregulation, hormones derived from adipose tissue, nutritional components, gut microbiota, and both genetic and epigenetic factors—play integral roles in its pathogenesis (Fig. 2) [17].
 have a profound impact on the pathogenesis of MASLD. (Created with BioRender.com). DNA, deoxyribonucleic acid; IRS, insulin receptor substrate; miRNA, microRNA; SNP, single nucleotide polymorphism.")
Various factors play integral roles in the pathogenesis of MASLD. Differential changes in multiple areas (including insulin resistance, genetics and epigenetics, gut microbiome, and lipid accumulation) have a profound impact on the pathogenesis of MASLD. (Created with BioRender.com). DNA, deoxyribonucleic acid; IRS, insulin receptor substrate; miRNA, microRNA; SNP, single nucleotide polymorphism.
Insulin resistance is a physiological state that refers to the disorder of molecular signaling pathways when insulin acts on target tissues, resulting in a decrease in the ability of cells to respond to insulin. Insulin resistance plays a key role in the development of MASLD. The reduced expression of insulin receptor substrate 2 during insulin resistance leads to the overexpression of sterol regulatory element-binding protein 1c, a transcription factor that promotes hepatic de novo lipogenesis (DNL), thereby upregulating DNL [18,19]. Moreover, the inhibited β-oxidation of free fatty acids (FFA) in liver cells causes further lipid accumulation [20]. Elevated intracellular FFA concentrations activate multiple serine kinases, including c-Jun N-terminal kinase (JNK), an inhibitor of nuclear factor κ-B kinase, and protein kinase C-θ [21], which phosphorylate serine residues of insulin receptor substrate 1, thereby reducing its activity and exacerbating insulin resistance [22]. Insulin resistance is the main feature of MASLD, and the two are in a mutually promoting bidirectional relationship.
2.2Hepatic lipid accumulationIn MASLD, lipid accumulation significantly affects liver health. In the liver, fatty acids are metabolized by mitochondrial β-oxidation or esterified to form triglycerides. Research indicates that as MASLD severity increases, there is a significant downregulation of both complete oxidation of long-chain fatty acids and the rate-limiting steps of β-oxidation in the liver. This leads to the excessive formation of triglyceride-laden lipid droplets, which is a hallmark feature of MASLD [23]. At the same time, the breakdown of these lipid droplets increases intracellular fatty acid levels. Specific lipotoxic lipids such as diacylglycerols, ceramides, and lysophosphatidylcholine may cause cell damage and trigger NASH. Lipotoxic lipids can induce pathological changes such as endoplasmic reticulum stress, inflammation, apoptosis, and injury responses. External factors like cytokine imbalance, ATP depletion, and periodic hypoxia further exacerbate hepatocyte lipotoxic stress and inflammation [24].
2.3Immune dysregulationThe liver, as a complex immunological organ, contains a variety of immune cell types, including Kupffer cells, non-Kupffer macrophages, natural killer (NK) cells, natural killer T (NKT) cells, dendritic cells (DCs), and hepatic stellate cells (HSCs), among others. Through their interactions, these cells play a critical role in maintaining hepatic immune homeostasis, defending against pathogen invasion, and regulating the progression of liver damage [25].
In MASLD, the polarization mechanisms of M1 and M2 macrophages are mediated through multiple signaling pathways. The polarization of M1 macrophages is primarily induced by pro-inflammatory factors such as saturated fatty acids (SFA) and lipopolysaccharides (LPS), which activate the NF-κB signaling pathway. This activation leads to the upregulation of pro-inflammatory cytokines, such as TNF-α and IL-6, and is mainly dependent on the activation of the TLR4 receptor [26,27]. In contrast, the polarization of M2 macrophages is mediated by anti-inflammatory factors such as n-3 polyunsaturated fatty acids (PUFA) and IL-4, which activate the PPAR-γ signaling pathway. This activation promotes the expression of anti-inflammatory cytokines, such as IL-10, and effectively inhibits the activity of the NF-κB signaling pathway [28,29]. Moreover, the activation of PPAR-γ can inhibit M1 polarization by directly binding to the NF-κB p65 subunit, thereby promoting M2 polarization. These mechanisms reveal the dynamic changes in M1 and M2 macrophage polarization and their roles in the progression of NAFLD [29].
2.4Gut microbiotaIn MASLD, poor dietary habits can induce a series of chain reactions by disrupting the gut microbiota, leading to dysbiosis, intestinal barrier disruption, and increased intestinal permeability, thereby allowing more pathogen-associated molecular patterns and bacteria to translocate through the portal vein into the liver, resulting in sustained immune activation and inflammation, which ultimately promotes the progression of MASLD [30]. Studies have shown that the gut microbiota can produce substantial amounts of ethanol in MASLD patients. This ethanol enters the liver through the portal circulation, bypassing the first-pass effect, thereby inducing hepatic steatosis and inflammation [31].
2.5Genetic and epigenetic factorsMASLD exhibits familial aggregation. In one study, 16 out of 90 MASLD patients had first-degree relatives who were also affected by the disease [32]. Furthermore, parents with type 2 diabetes have an increased risk of MASLD in their offspring [33]. Research on obese children with MASLD and their family members revealed that siblings and parents of MASLD children have a higher probability of developing fatty liver, with a stronger correlation between liver fat fraction and body mass index (BMI) [34]. Twin studies have shown that after adjusting for gender and BMI, 60 % of the variation in serum alanine aminotransferase (ALT), a marker of liver damage (hepatitis, cirrhosis, fatty liver), is determined by genetic factors [35].
Although MASLD is defined by its histological phenotype, genome-wide association studies have identified several strongly associated genes. The PNPLA3 gene plays a pivotal role in various aspects of MASLD. Studies indicate that the PNPLA3 rs738409 (Ile148Met) variant is associated with increased inflammation and fibrosis in MASLD [33,36,37]. This variant also elevates the risk of MASLD-associated HCC [37]. The PNPLA3 gene modulates hepatic steatosis by enhancing the liver's response to environmental stressors [37,38]. The GCKR rs1260326 (Pro446Leu) variant leads to increased hepatic glucokinase activity, perpetuating hepatic glycolysis, downregulating glucose and insulin levels, increasing malonyl-CoA content in hepatocytes, inhibiting fatty acid β-oxidation, and promoting lipid accumulation [38]. The TM6SF2 rs58542926 (Glu167Lys) variant results in loss of protein function, leading to decreased VLDL levels, elevated ALT levels, and hepatic steatosis [39] . Genes such as superoxide dismutase 2, phosphatidylethanolamine N-methyltransferase, angiotensin II receptor type 1, and kruppel-like factor 6 have been found to be independently associated with MASLD progression [40–43].
During the progression from simple fatty liver to NASH, epigenetic modifications such as DNA methylation, histone acetylation, and miRNAs play crucial roles [44]. The DNA methylation levels of apolipoprotein B and nuclear factor erythroid 2–related factor 2 are positively correlated with lipid accumulation in MASLD, whereas the methylation level of dipeptidyl peptidase 4 is negatively correlated with NASH staging [45–47]. The SIRT protein family and specific miRNAs (such as miR-122) significantly influence the development of NASH by regulating key genes involved in lipid and glucose metabolism [41,48]. Furthermore, these genetic modifications can be inherited by offspring, increasing the risk of MASLD [49]. Small RNAs derived from the 5′ end of tRNA, known as tsRNA, have been found to be significantly upregulated in the sperm tsRNA of high-fat diet (HFD) mice, with increased m5C and m2G modifications. Changes in the paternal RNA modification profile can be transmitted to offspring, thereby perpetuating metabolic disorder traits [50].
3Overview of mitochondria3.1Structure and dynamics of mitochondriaMitochondria are double-membrane organelles found in most eukaryotic cells, consisting of several functional regions including the outer membrane, intermembrane space, inner membrane, and matrix [51]. They have their own double-stranded circular DNA, independent of nuclear DNA, and are able to replicate, transcribe, and translate mitochondrial-specific proteins. These proteins include 13 polypeptides of the mitochondrial respiratory chain, 22 transfer RNAs, and 2 ribosomal RNAs, all of which are essential for mitochondrial function and self-replication [52].
Mitochondrial dynamics refer to the continuous processes of mitochondrial fusion and fission, regulated by specific proteins, which maintain mitochondrial homeostasis [53]. Mitochondrial fission involves multiple steps: first, actin filaments assist the endoplasmic reticulum in tightly wrapping around the mitochondrion, leading to local constriction of the mitochondrial surface and providing a platform for the assembly of dynamin-related protein 1 (DRP1) [54,55]; Next, DRP1 mediates further constriction, promoting the recruitment of dynamin 2 (DNM2); finally, mitochondrial division is completed through membrane scission induced by DNM2 [56].
Oxidative phosphorylation (OXPHOS) is a vital mitochondrial function involving multiple enzyme complexes in the inner mitochondrial membrane, which generate ATP by transferring electrons from NADH and FADH2 [57,58]. During this process, electrons sequentially pass through complexes I, III, and IV, ultimately combining with oxygen and yielding H2O [59]. This electron transfer drives the translocation of protons from the mitochondrial matrix across the membrane, creating a proton motive force. This force, consisting of an electrochemical gradient, powers ATP synthase to synthesize ATP [60]. However, electron leakage in the electron transport chain can lead to the generation of ROS, which serves as signaling molecules in cells, though excessive ROS can cause oxidative damage.
Mitochondrial redox homeostasis affects mitochondrial dynamics through redox-sensitive post-translational modifications of specific proteins. An illustration of this is the enhancement of Drp1′s GTPase activity via S-nitrosylation at Cys644, thereby promoting mitochondrial fission [61]. S-nitrosylation of optic atrophy 1 (OPA1) may be involved in mitochondrial fusion, although the specific function is still unclear [62]. Mitofusin 2(Mfn2) is susceptible to ubiquitination and degradation after JNK-mediated phosphorylation, leading to mitochondrial fission [63]. Additionally, S-nitrosylation of Parkin inhibits its E3 ubiquitin ligase activity, affecting the degradation of Mfn1 and possibly promoting mitochondrial elongation [64]. These proteins interact with each other and, in conjunction with changes in redox status, dynamically regulate mitochondrial morphology.
3.2Mitochondrial dysfunctionMitochondrial dysfunction includes a variety of damages, such as reduced ATP production due to ETC damage, ROS generation, calcium ion dysregulation, increased mitochondrial membrane permeability due to the opening of the permeability transition pore (PTP), and dysregulation of cell apoptosis. mtDNA, as the energy metabolism center of the cell, is more susceptible to damage by toxic metabolites. Compared with nuclear DNA, mtDNA lacks effective protection and repair mechanisms and is more susceptible to damage and mutation [65]. Damage to mtDNA impairs mitochondrial function, reduces energy production, and leads to a variety of diseases, including mtDNA depletion syndrome, multiple cancers, and age-related diseases [66–71]. PTP is a voltage-dependent channel protein complex located in the inner mitochondrial membrane. When PTP is open, mitochondrial membrane permeability increases significantly, allowing small molecules such as water, inorganic ions, metabolites, and proteins to pass through. High levels of ROS, Ca2+ overload, changes in the lipid microenvironment, inactivation of B-cell lymphoma 2 protein, and high pH environment can trigger the opening of PTP [72,73], ultimately leading to mitochondrial swelling and membrane rupture. This process releases various apoptotic factors, such as cytochrome c, which activate downstream effectors like caspases, resulting in abnormal cell apoptosis [74].
4Mitochondrial dysfunction and MASLDMitochondria play a crucial role in the development of MASLD by regulating fatty acid β-oxidation, managing oxidative stress, and interacting with cellular structures such as lipid droplets and the endoplasmic reticulum. Dysfunction in these processes leads to increased ROS production, mtDNA damage, and impaired mitophagy, which contribute to hepatic steatosis, inflammation, and fibrosis. Understanding these mitochondrial mechanisms is essential for developing targeted therapeutic strategies to mitigate the progression of MASLD (Fig. 3).
. ACSL1, acyl-CoA synthetase long-chain family member 1; ACSL4, acyl-CoA synthetase long-chain family member 4; ADP, adenosine diphosphate; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; ATP synthase, adenosine triphosphate synthase; CD36, cluster of differentiation 36; CPT, carnitine palmitoyltransferase; ETC damage, electron transport chain damage; FADH, flavin adenine dinucleotide (Reduced Form); MASLD, metabolic dysfunction-associated steatotic liver disease; MAM, mitochondria-associated membranes; MFN2, mitofusin 2; mtDNA, mitochondrial DNA; NADH, nicotinamide adenine dinucleotide (Reduced Form); PI3K/AKT, phosphoinositide 3-kinase / protein kinase B pathway; ROS, reactive oxygen species; SMAD3, SMAD family member 3; ULK1, unc-51 like autophagy activating kinase 1.")
Pathophysiological mechanisms by which Mitochondrial Dysfunction Affects MASLD. CD36 regulates transport proteins that transport fatty acids into cells. CD36 can promote the transport of long-chain fatty acids into the cell via transport proteins, and palmitoylated CD36 interacts with ACSL1 to inhibit β-oxidation. ACSL4 can also regulate PGC1α in a smad3-dependent manner to affect β-oxidation. These processes result in the accumulation of fatty acids within the hepatocyte. Impaired ETC and increased ROS production lead to lipotoxicity, which in turn damages mtDNA and exacerbates changes in mitochondrial structure and function. The accumulation of lipotoxicity and the inhibition of the PI3K/AKT and AMPK pathways impair mitochondrial autophagy. In addition, decreased MFN2 expression in MAMs leads to reduced phosphatidylcholine synthesis. This series of events ultimately promotes hepatic steatosis, inflammation, and fibrosis. (Created with BioRender.com). ACSL1, acyl-CoA synthetase long-chain family member 1; ACSL4, acyl-CoA synthetase long-chain family member 4; ADP, adenosine diphosphate; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; ATP synthase, adenosine triphosphate synthase; CD36, cluster of differentiation 36; CPT, carnitine palmitoyltransferase; ETC damage, electron transport chain damage; FADH, flavin adenine dinucleotide (Reduced Form); MASLD, metabolic dysfunction-associated steatotic liver disease; MAM, mitochondria-associated membranes; MFN2, mitofusin 2; mtDNA, mitochondrial DNA; NADH, nicotinamide adenine dinucleotide (Reduced Form); PI3K/AKT, phosphoinositide 3-kinase / protein kinase B pathway; ROS, reactive oxygen species; SMAD3, SMAD family member 3; ULK1, unc-51 like autophagy activating kinase 1.
Mitochondrial fatty acid β-oxidation is the primary pathway for fatty acid degradation and is crucial for maintaining energy homeostasis in the human body. During the post-absorptive state and fasting, fatty acids become a key energy source due to limited glucose availability. However, even in glucose-rich conditions, fatty acid β-oxidation remains a major energy source for the heart, skeletal muscles, and kidneys. In the cytoplasm, fatty acids are activated into acyl-CoA by acyl-CoA synthetase and then transported into the mitochondria via the carnitine shuttle. Subsequently, acyl-CoA undergoes four enzymatic steps: dehydrogenation, hydration, re-dehydrogenation, and thiolysis, being broken down into acetyl-CoA, with the generated FADH2 and NADH entering the ETC [75].
In the early stages of insulin resistance-related NASH, the liver responds to lipid overload by increasing β-oxidation activity and VLDL secretion [76]. Insulin resistance enhances lipolysis in adipose tissue, releasing a large amount of FFA into the bloodstream, which are subsequently absorbed by the liver. As β-oxidation increases, a substantial amount of ROS is generated, leading to oxidative stress and mitochondrial damage. Persistent oxidative stress and mitochondrial damage gradually impair β-oxidation function, reducing the liverʼs efficiency in processing FFA, resulting in further lipid accumulation and forming a positive feedback loop of hepatocellular damage in MASLD [77]. When hepatic β-oxidation is obstructed, fatty acids produce lipotoxic substances that induce pro-inflammatory and pro-fibrotic signaling pathways, promoting steatohepatitis and fibrosis. Reversing the inhibition of β-oxidation can slow the progression of MASLD [78]. Cluster of differentiation 36 (CD36) facilitates the uptake of long-chain fatty acids by hepatocytes. In MASLD, palmitoylation of CD36 is significantly increased. Depalmitoylation of CD36 increases its mitochondrial distribution and, through interaction with long-chain acyl-CoA synthetase 1, upregulates mitochondrial β-oxidation, thereby reducing lipid accumulation in hepatocytes [79]. The long-chain acyl-CoA synthetase family member ACSL4 is highly expressed in the liver tissues of MASLD patients and is positively correlated with hepatocellular steatosis and fibrosis. Although ACSL4 does not directly participate in β-oxidation, its silencing upregulates β-oxidation-related enzymes peroxisome proliferator-activated receptor α and its coactivator peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) in an SMAD family member 3-dependent manner, promoting mitochondrial β-oxidation and thereby protecting cells from death [80].
4.2Mitochondrial oxidative stress and MASLDMitochondrial oxidative stress is characterized by excessive production and accumulation of ROS due to mitochondrial dysfunction, leading to cellular oxidative damage. Under normal metabolic conditions, ROS are produced by the ETC, but excessive ROS can oxidize proteins, lipids, and DNA, resulting in cellular dysfunction and death [81]. Enzymatic antioxidants such as superoxide dismutase and catalase, along with non-enzymatic antioxidants like glutathione and ascorbic acid, work together to eliminate ROS. However, when this balance is disrupted, oxidative stress intensifies, influencing the onset and progression of MASLD [10,11]. Mitochondrial antioxidant capacity is influenced by several redox pairs, including NADH/NAD+ and reduced glutathione /oxidized glutathione. Changes in the mitochondrial redox state lead to an increased NADH/NAD+ ratio, which activates fatty acid synthases (such as fatty acid synthase and glycerol-3-phosphate acyltransferase) and inhibits fatty acid oxidases (such as acyl-CoA dehydrogenase and β-hydroxyacyl-CoA dehydrogenase) [82]. As MASLD progresses, fatty acids accumulate in the liver. To control excess lipids, hepatocytes enhance fatty acid oxidation and the tricarboxylic acid (TCA) cycle [83]. However, lipid oxidation produces a large amount of reducing equivalents, leading to electron overload of the mitochondrial respiratory chain, thereby increasing the production of ROS [84].
ROS not only induce the directed migration of pro-fibrotic cells in the liver, promoting hepatic fibrosis, but also trigger lipid peroxidation of PUFAs, producing harmful aldehyde byproducts such as 4‑hydroxy-2-nonenal and malondialdehyde. This exacerbates cellular damage and leads to cell death [85]. Furthermore, due to insufficient mitochondrial antioxidant defense mechanisms to cope with sustained oxidative stress induced by long-chain free fatty acids, the mitochondrial ETC and mtDNA suffer oxidative damage. This damage not only impairs normal mitochondrial function but also promotes the generation of more ROS, creating a vicious cycle that accelerates the progression of MASLD [86–88]. The methylation-controlled J protein (MCJ) is a transmembrane protein located in the mitochondrial inner membrane, possessing a unique regulatory mechanism for fatty acid metabolism. As a negative regulator of ETC complex I, MCJ inhibits its expression, which can enhance the activity of mitochondrial complex I, thereby increasing the efficiency of fatty acid β-oxidation. Inhibiting MCJ not only boosts the activity of complex I but also promotes the formation of respiratory chain complexes I, III, and IV, reducing electron leakage, thereby preventing ROS production and the downstream activation of the JNK pathway, ultimately alleviating MASLD [89].
4.3Peridroplet mitochondria and MASLDPeridroplet Mitochondria (PDM) are mitochondria closely arranged around lipid droplets, playing an important role in cellular energy and lipid metabolism. PDM enhance fatty acid β-oxidation and ATP synthesis, providing necessary energy for cellular metabolic activities, and support triglyceride synthesis and the maintenance of lipid homeostasis. Additionally, PDM capture free fatty acids, reducing their accumulation within cells, thereby decreasing lipotoxicity and protecting cells from damage caused by lipid overload. These functions make PDM crucial in regulating cellular metabolism [90]. In contrast, cytosolic mitochondria (CM) mainly regulate cellular energy metabolism through the transport and oxidation of fatty acids, complementing the role of PDM in lipid metabolism.
In the pathological mechanisms of MASLD, abnormal accumulation of hepatic lipid droplets and mitochondrial dysfunction are key features. PLIN5, as a lipid droplet-associated protein, regulates PDM, promoting fatty acid release and oxidative metabolism, thereby reducing lipid droplet accumulation and lipotoxicity, and maintaining the balance of hepatic lipid metabolism [91,92]. Additionally, PLIN5 plays a dual role in lipolysis and lipid metabolism, depending on the cell's energy status and hormonal environment. After catecholamine stimulation, PLIN5 is phosphorylated and translocates to the nucleus, where it forms a complex with SIRT1 and PGC-1α. By activating the deacetylase activity of SIRT1, PLIN5 enhances the deacetylation of PGC-1α, thereby promoting the expression of PGC-1α-regulated fatty acid oxidation genes. This mechanism ensures that the fatty acids released during lipolysis are efficiently utilized by mitochondria, avoiding mitochondrial overload and maintaining their function and metabolic efficiency [93]. Recent research by Xiangyun Sun et al. has shown that diethyldithiocarbamate (DDC) can upregulate the expression of PLIN5, thereby promoting the formation of PDM. DDC improves mitochondrial function by enhancing mitochondrial OXPHOS levels and increasing ATP production. This mechanism not only promotes the synthesis of triglycerides (TG) but also significantly alleviates mitochondrial dysfunction caused by MASLD, reducing liver inflammation and fibrosis. Consequently, it inhibits the progression of MASLD to more severe stages, such as metabolic-associated steatohepatitis (MASH) [94].
4.4Mitophagy and MASLDMitophagy refers to the selective degradation of damaged and dysfunctional mitochondria by the cell, which helps maintain the normal structure, quantity, and function of mitochondria [95]. In the liver, there are two main pathways of mitochondrial autophagy: ubiquitin-dependent pathway and ubiquitin-independent pathway. In the ubiquitin-dependent pathway, E3 ubiquitin ligases such as Parkin bind ubiquitin to mitochondrial outer membrane proteins and recruit autophagy receptors such as p62, OPTN and NDP52. These autophagy receptors contain Microtubule-associated protein 1 light chain 3 (LC3) interacting regions (LIRs), which can bind to LC3 and anchor ubiquitinated mitochondria to the autophagosome membrane, leading to the degradation of damaged mitochondria. In the ubiquitin-independent pathway, receptors on the mitochondrial outer membrane, such as FUN14 domain-containing protein 1, contain LIR sequences and can directly bind to LC3 under stress conditions to form autophagosome membranes that encapsulate damaged mitochondria and complete mitophagy [96]. Studies have shown that upregulation of macrophage stimulating factor 1 (Mst1) leads to liver vacuolation, steatosis, fibrosis, oxidative stress, and inflammation, and plays a key role in HFD-induced MASLD. Mst1 gene knockout alleviates HFD-induced liver damage by activating the AMP-activated protein kinase (AMPK) signaling pathway, regulating Parkin-related mitophagy [97]. Additionally, as a key protein in glucose regulation, AMPK also regulates mitophagy through the ubiquitin-independent pathway by activating the autophagy-related protein Unc-51-like kinase 1 [98].
In MASLD, an imbalance between mitochondrial fusion and fission leads to abnormally enlarged mitochondria, affecting the mitophagy process. DRP1 and OPA1 respectively regulate mitochondrial fission and fusion in hepatocytes, with their actions being antagonistic to each other. Research has found that simultaneous knockout of DRP1 and OPA1 in mice maintains mitochondrial morphology stability similar to that of the control group. In MASLD model mice, OPA1 knockout significantly reduces mitochondrial size, thereby promoting mitophagy and preventing the progression of MASLD [99].
4.5mt DNA and MASLDPatients with MASLD exhibit a higher mutation rate in mitochondrial DNA (mt DNA) compared to healthy individuals [14]. Studies have identified that the progression of MASLD is closely related to nucleotide variations in the d-loop region of mt DNA. Specifically, the m.16318C>A mutation is associated with NASH, while the Mt16129AA homozygous mutation genotype correlates with a higher degree of liver fibrosis [100]. In another cohort of MASLD patients, the mtDNA mutation profile in the liver includes several point mutations related to the disease state. These mutations primarily target the OXPHOS system and affect phenotypes. Notably, the m.14766 C > T missense mutation in the mitochondrial cytochrome b (MT-CYB) gene is predicted to be a deleterious mutation. MASLD patients carrying this mutation exhibit significant mitochondrial morphological changes under electron microscopy, including increased mitochondrial volume, condensed mitochondrial matrix, loss of mitochondrial membranes and cristae, and peroxisome proliferation [14]. Additionally, studies have indicated that MT-CYB mutations are associated with histological features such as steatosis, inflammation, and fibrosis. Lipid peroxidation and ROS are suggested as the causes of MT-CYB mutations [101].
4.6Interactions between mitochondria and endoplasmic reticulum in MASLDIn 1971, Franke et al. observed a close association between the mitochondrial outer membrane and the endoplasmic reticulum (ER) in vivo [102]. Mitochondria-associated endoplasmic reticulum membranes (MAM) are structures rich in phospholipid-synthesizing enzymes such as diacylglycerol acyltransferase, phosphatidylserine synthase, and cholesterol acyltransferase, primarily responsible for lipid transport between the ER and mitochondria [103]. MAMs contain many proteins, including inositol 1,4,5-triphosphate receptor (IP3R), voltage-dependent anion channel (VDAC), Sigma-1 receptor, glucose-regulated protein 75, and various calcium-binding partners. These proteins mediate efficient calcium ion transfer from the ER to mitochondria, participating in the regulation of cellular energy metabolism, survival, and death processes [104].
In MASLD, loss of CDP-diacylglycerol synthase 2 (CDS2) results in reduced mitochondrial content in the liver and reduced activity of ETC complexes I and II, along with triglyceride Ester, cholesterol, ALT and AST levels are increased. Furthermore, reduced CDS2 expression altered the protein composition of MAMs, including significant downregulation of the mitochondrial protein Mitofusin 2 (MFN2). [105]. MFN2 is a mitochondrial membrane protein that connects mitochondria and the ER membrane, selectively binding and separating phosphatidylserine (PS) from the membrane, forming PS-rich rigid domains. Reduced MFN2 expression in MASLD leads to decreased PS transport from the ER to mitochondria, thereby inhibiting the synthesis of phosphatidylcholine in mitochondria. Disruption of phospholipid metabolism-induced hepatic MAM remodeling has been shown to result in triglyceride accumulation and insulin resistance, thereby promoting the progression of MASLD [106].
With industrial development, the impact of environmental factors on the progression of MASLD has become increasingly significant. In mice exposed to Nanoplastic (NP), a common environmental pollutant, mitochondrial swelling, cristae disappearance, endoplasmic reticulum dilation, and more frequent contacts between mitochondria and the endoplasmic reticulum were observed, resulting in the formation of more MAMs. Additionally, NP increases the levels of IP3R1, GRP75, and VDAC1 in MAMs by binding to IP3R1, enhancing the stability of these proteins and promoting MAM formation. The increased MAMs enhance the uptake of Ca2+ by mitochondria, leading to mitochondrial Ca2+ overload and excessive production of ROS, which suppresses downstream NRF2. By binding to the promoter region of the microRNA-26a (miR26a) gene, downregulated NRF2 inhibits the expression of miR26a, which promotes the expression of the target gene VDAC1 and facilitates the nuclear translocation of nuclear factor p65 (p65) and kelch-like ECH-associated protein 1, leading to the inactivation of NRF2. Downregulation of miR26a forms a positive feedback loop that exacerbates oxidative stress, steatosis, inflammation, and fibrosis in the liver, thereby promoting the progression of MASLD [107].
5Mitochondrial dysfunction: a therapeutic target for MASLDRecent clinical studies have evaluated the impact of dietary interventions and bariatric surgery on hepatic mitochondria, which are emerging as a promising therapeutic target for MASLD.
Numerous studies have demonstrated that enhancing hepatic mitochondrial β-oxidation through non-pharmacological treatments can reduce the accumulation of free fatty acids, thereby alleviating hepatic steatosis and inflammation. For instance, glycine treatment has been shown to enhance mitochondrial activity and reduce lipid accumulation [108]. Regular physical activity is another effective non-pharmacological intervention, as it improves the pathological characteristics of NAFLD by enhancing hepatic mitochondrial fatty acid oxidation and modulating the structure of PTP, thus reducing mitochondrial damage [109]. Caloric restriction is an effective strategy for treating metabolic disorders. The ketogenic diet can significantly improve liver fatty acid content, and metabolic surgery has been shown to significantly affect patient weight, increase OXPHOS capacity, enhance citrate synthase activity, promote ETC gene expression, and reduce oxidative stress [110,111].
In the pharmacological treatment of MASLD, pioglitazone ameliorates insulin resistance by increasing mitochondrial activity and reducing lipid accumulation [112]. Vitamin E alleviates mitochondrial dysfunction through its powerful antioxidant capacity. It can scavenge ROS and reactive nitrogen species (RNS) to protect mitochondria from oxidative damage [113]. In addition, vitamin E enhances the activities of antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase, enhancing cellular antioxidant defense [114–116]. Vitamin E also improves mitochondrial membrane potential by increasing the level of anti-apoptotic protein BCL-2, reducing the level of pro-apoptotic proteins BAX and p53, and inhibiting caspase activity, thereby reducing cell apoptosis [117]. In March 2024, the U.S. Food and Drug Administration (FDA) approved resmetirom to treat MASH with moderate to advanced fibrosis for the first time. Its derivative, HSK31679, is currently undergoing Phase II clinical trials [118,119]. Resmetirom alleviates MASH by targeting the liver-specific thyroid hormone receptor β, stimulating hepatic fatty acid β-oxidation, and thereby reducing the burden of lipotoxic lipids [120]. Additionally, Resmetirom decreases oxidative stress and inflammation, lowering the risk of fibrosis, and promotes mitochondrial biogenesis to maintain mitochondrial health. For example, in cultured HepG2–THR-β cells from mouse models, thyroid hormone treatment increased mitochondrial respiration and fatty acid oxidation under both basal and palmitic acid-treated conditions, while reducing inflammation and fibrotic responses induced by lipopolysaccharides and palmitic acid stimulation [121–123]. Cotadutide is a novel dual agonist that activates both the glucagon-like peptide-1 receptor and the glucagon receptor (GCGR), exerting its therapeutic effects by inhibiting fatty acid synthesis through GCGR-mediated pathways. In addition, it promotes mitochondrial turnover, facilitates the maintenance of mitochondrial function, and alleviates hepatic steatosis, phosphorylation, and inflammation [124]. Another dual agonist, survodutide, enhances gluconeogenesis and glycogenolysis while reducing hepatic lipid accumulation. It also supports mitochondrial renewal and reduces oxidative stress, further demonstrating its potential in metabolic and liver disease interventions [125]. Recent study has found that endogenous unfolded albumin in the cytoplasm undergoes phase separation to form membrane-less, shell-like organelles called proteosomes. Carnitine palmitoyltransferase 2 (CPT2) transports fatty acids from the mitochondrial outer membrane into the matrix for β-oxidation, while proteosomes capture CPT2 and regulate its entry into mitochondria, thereby modulating mitochondrial metabolism and respiration. The Hsp90 inhibitor 17-Allylamino-17-demethoxygeldanamycin (17-AAG) can promote the accumulation of hepatic proteosomes, thereby inhibiting the progression of NAFLD, reducing mitochondrial oxidative stress in the early stages of MASLD, and decreasing ROS production [126].
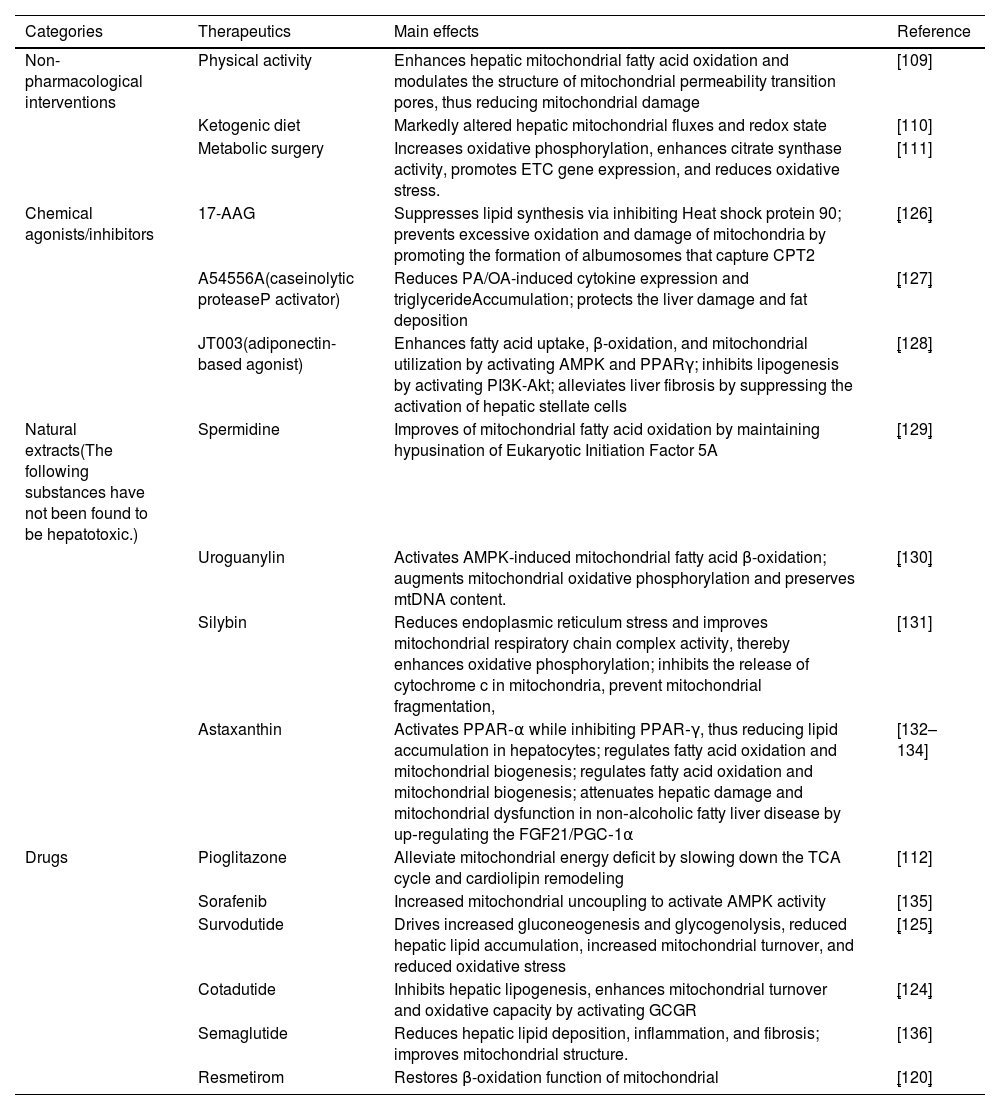
In summary, mitochondrial dysfunction represents a promising target for the treatment of MASLD. The combination of non-pharmacological and pharmacological interventions, including increased physical activity, dietary modifications, anti-diabetic medications, and thyroxine analogs, has been shown to significantly enhance mitochondrial function, thereby alleviating the symptoms of MASLD and slowing its progression (Table 1).
Therapeutic intervention for mitochondria in MASLD.
| Categories | Therapeutics | Main effects | Reference |
|---|---|---|---|
| Non-pharmacological interventions | Physical activity | Enhances hepatic mitochondrial fatty acid oxidation and modulates the structure of mitochondrial permeability transition pores, thus reducing mitochondrial damage | [109] |
| Ketogenic diet | Markedly altered hepatic mitochondrial fluxes and redox state | [110] | |
| Metabolic surgery | Increases oxidative phosphorylation, enhances citrate synthase activity, promotes ETC gene expression, and reduces oxidative stress. | [111] | |
| Chemical agonists/inhibitors | 17-AAG | Suppresses lipid synthesis via inhibiting Heat shock protein 90; prevents excessive oxidation and damage of mitochondria by promoting the formation of albumosomes that capture CPT2 | [126] |
| A54556A(caseinolytic proteaseP activator) | Reduces PA/OA-induced cytokine expression and triglycerideAccumulation; protects the liver damage and fat deposition | [127] | |
| JT003(adiponectin-based agonist) | Enhances fatty acid uptake, β-oxidation, and mitochondrial utilization by activating AMPK and PPARγ; inhibits lipogenesis by activating PI3K-Akt; alleviates liver fibrosis by suppressing the activation of hepatic stellate cells | [128] | |
| Natural extracts(The following substances have not been found to be hepatotoxic.) | Spermidine | Improves of mitochondrial fatty acid oxidation by maintaining hypusination of Eukaryotic Initiation Factor 5A | [129] |
| Uroguanylin | Activates AMPK-induced mitochondrial fatty acid β-oxidation; augments mitochondrial oxidative phosphorylation and preserves mtDNA content. | [130] | |
| Silybin | Reduces endoplasmic reticulum stress and improves mitochondrial respiratory chain complex activity, thereby enhances oxidative phosphorylation; inhibits the release of cytochrome c in mitochondria, prevent mitochondrial fragmentation, | [131] | |
| Astaxanthin | Activates PPAR‐α while inhibiting PPAR‐γ, thus reducing lipid accumulation in hepatocytes; regulates fatty acid oxidation and mitochondrial biogenesis; regulates fatty acid oxidation and mitochondrial biogenesis; attenuates hepatic damage and mitochondrial dysfunction in non‐alcoholic fatty liver disease by up‐regulating the FGF21/PGC‐1α | [132–134] | |
| Drugs | Pioglitazone | Alleviate mitochondrial energy deficit by slowing down the TCA cycle and cardiolipin remodeling | [112] |
| Sorafenib | Increased mitochondrial uncoupling to activate AMPK activity | [135] | |
| Survodutide | Drives increased gluconeogenesis and glycogenolysis, reduced hepatic lipid accumulation, increased mitochondrial turnover, and reduced oxidative stress | [125] | |
| Cotadutide | Inhibits hepatic lipogenesis, enhances mitochondrial turnover and oxidative capacity by activating GCGR | [124] | |
| Semaglutide | Reduces hepatic lipid deposition, inflammation, and fibrosis; improves mitochondrial structure. | [136] | |
| Resmetirom | Restores β-oxidation function of mitochondrial | [120] |
MASLD, a highly prevalent liver disease, has a complex and multifaceted pathogenesis involving insulin resistance, immune response, gut microbiota, and genetic and epigenetic factors, among others. Mitochondrial dysfunction plays a crucial role in the occurrence and development of MASLD, including impaired β-oxidation, PDM, oxidative stress, defective mitophagy, and mtDNA mutations (Fig. 3). The reduction in fatty acid oxidation and mitochondrial enzyme activity precedes the onset of insulin resistance and MASLD, with decreased insulin sensitivity and diminished levels of free radical scavenging enzymes such as superoxide dismutase exacerbating the loss of mitochondrial function [137]. In recent years, research into the relationship between mitochondria and MASLD has deepened, revealing how mitochondrial dysfunction accelerates MASLD progression by affecting hepatocyte metabolism, oxidative stress, and apoptosis. Particularly, the interaction between mitochondria and the endoplasmic reticulum in lipid metabolism regulation underscores the central role of mitochondria in the pathogenesis of MASLD. In the future, diagnostic and therapeutic strategies targeting mitochondrial dysfunction are expected to become new directions for clinical intervention in MASLD, providing more treatment options and better prognoses for patients.
Authors contributionsChenyang Mu conceived and wrote the manuscript. Sijie Wang and Fu Yang conceived and revised the manuscript. All authors collected and analyzed the data.
FundingThis article was supported by grants from the Noncommunicable Chronic Diseases-National Science and Technology Major Project (2023ZD0500102) and the National Natural Science Foundation of China (No. 82472780).
Availability of data and materialsNo applicable.
The figures of this review were created by Biorender.



