






Background: Hepatocyte growth factor (HGF) is not only an antiapoptotic and antifibrotic factor of liver, but it is also an adipokine. Serum HGF levels are strongly associated with liver diseases, obesity, insulin resistance (IR), and metabolic syndrome (MS). Non-alcoholic steatohepatitis (NASH) is the hepatic component of MS. To the best of our knowledge, serum HGF levels in patients with NASH have not been previously studied. Our aim was to elucidate the correlation of HGF with the clinical and histopathological parameters of NASH. Methods: The study group consisted of 26 patients (13 men) who had clinical diagnoses of NASH and underwent liver biopsies. Controls were 13 volunteers (3 men) with negative viral autoimmune markers, and with normal levels of serum lipids and liver enzymes. Results: Among the NASH patients, 14 (54%) were overweight and 10 (39%) had grade I-II obesity. All the patients had class 3-4 non-alcoholic fatty liver disease (NAFLD) except for 2 who had class 2 disease. All of the patients had Child’s class A liver disease, and MS was present in 5 (19%) patients and 8 (31%) patients had Homeostasis Model Assessment of Insulin Resistance (HOMA) > 3. Serum HGF levels were similar in NASH patients (1.24 ± 1.09 pg/mL) and controls (0.86 ± 0.22 pg/mL) (p = 0.21). The levels of serum HGF did not differ between the patients with or without MS (1.65 ± 1.48 pg/mL and 1.04 ± 0.80 pg/mL, respectively, p=0.65). HGF was not correlated with the laboratory or histopathological parameters. Conclusions: Serum HGF levels were higher in NASH patients than in the controls, although it was statistically insignificant and a correlation with MS could not be detected in this study.
Non-alcoholic fatty liver disease (NAFLD) defines a spectrum of liver diseases in which fat-laden hepatocytes exceed more than 5-10% of the total number of hepatocytes. Class 1 NAFLD constitutes simple hepatosteatosis, and higher classes (class 3-4 and maybe class 2) are defined as non-alcoholic steatohepatitis (NASH).1,2 There are uncertainties about the natural prognosis of NAFLD. It is as yet unclear if early classes of NAFLD progress to NASH or if NASH is a de nova disease. However, early classes of NAFLD could be accepted as precursor lesions of NASH.3 NASH is considered the hepatic component of metabolic syndrome (MS),4-6 which makes the clinical diagnosis of insulin resistance (IR) possible. All of the predictors of progression of fibrosis in patients with NASH such as age (> 45 years old), obesity ([body mass index] BMI > 30 kg/m6), fibrosis or cirrhosis (AST/ALT > 1), and diabetes, are associated with increased IR.2,7 As a result, even though NASH is a multifactorial disease, IR seems to be the main factor that is responsible for the progression of simple steatosis to NASH.
Adipose tissue is considered an endocrine organ regulating body metabolism. Adipocytes not only store energy, but also respond to metabolic signals by secreting hormones and cytokines that exert effects locally (adipose tissue), centrally (neuronal tissue), and peripherally (organs such as liver, muscle, pancreas).7 Adipocytokines or adipokines are any of the substances released from adipose tissue.8 Multiple adipokines have been identified that have significant effect on whole-body homeostasis, eating behavior, and insulin sensitivity. The major adipokines are leptin, adiponectin, and resistin, while, several other molecules, including TNF α, IL-6, PAI, complement proteins, and proteins of the rennin-angiotensin system, also act as adipokines.7,10,11 Recently, it has been shown that adipokines act on hepatocytes and kuppfer cells and initiate fibrosis in the liver. Peripheral IR in NASH patients leads to increased transport of fatty acids from adipose tissue to the liver, thereby overloading the pathways of fat metabolism in hepatocytes. Together with increased oxidative stress and/or mitochondrial dysfunction in hepatocytes, adipokines launch a vicious cycle ending with the development of NASH.23,12,13
Hepatocyte growth factor (HGF) has an important function in embryonic development, in tissue regeneration during acute injury, and also in preventing tissue fibrosis during chronic injury.14,15 In addition to being a growth factor, HGF is also an adipokine.7,16 Recently, Hiratsuka et al. reported that several components of MS are associated with elevated serum levels of HGF, independent of liver disease.17 Although HGF is associated with acute and chronic regeneration of the liver as well as MS, to the best of our knowledge, serum HGF levels in patients with NASH has not been previously studied. Accordingly, the aim of this study was to evaluate HGF in patients with NASH in respect to the presence of IR and/or MS.
Patients and methodsTwenty-six patients with NASH and 13 controls were included in this study. The diagnosis of NASH was based on the persistent elevation of alanine transaminase (ALT) of > 1.3 times the upper normal value for 3 months in spite of life-style changes, and null or negligible (< 20 g/day) alcohol consumption. Additionally, patients had negative hepatitis B and C markers and autoantibodies (antinuclear antibody, antismooth muscle antibody, liver-kidney-microsomal antibody, and antimitochondrial antibody), normal serum ceruloplasmin and α1 antitrypsin levels, and urinary copper excretion. Patients with diabetes or being treated with hepatotoxic drugs were excluded from the study. The patients with persistent hypertran-saminasemia after 3 months of life style changes underwent ultrasonography guided liver biopsies. All the liver biopsies were examined by the same experienced pathologist according to the criteria proposed by Brunt.17 Controls were chosen among patients with functional gastrointestinal disorders who had normal weight, blood pressure, fasting blood glucose, liver enzymes, and lipid parameters.
Body weight and height were measured to the nearest half-kilogram and BMI was calculated as weight (kilograms) divided by height squared (square meters). Patients with a BMI between 25 and 30 kg/m2 were considered over-weight and those with a BMI greater than or equal to 30 kg/m2 were considered obese. Waist circumference was measured at the smallest circumference between the point below the lower rib margin and the iliac crest.
Fasting blood samples of the patients were analyzed for glucose, insulin, c-peptide, lipid parameters, and liver enzymes. HGF measurement was performed with a commercially available ELISA kit (Quantikine HGF Immunoassay Kit, California, USA).
We identified IR by using the 75 mg Oral Glucose Tolerance Test (OGTT categorized according to 2nd hour postload glucose levels: normal was < 140 mg/dL, impaired was 140-200 mg/dL, and diabetic was > 200 mg/dL) and Homeostasis Model Assessment of Insulin Resistance (HOMA, it was calculated as insulin (mU/L) × glucose (µmol/L)/22.5). HOMA values equal to or greater than 3.0 were considered to be indicative of IR. We used the diagnostic criteria of the National Cholesterol Education Program Adult Treatment Panel III (ATP III) for the definition of MS. Diagnosis was made if 3 or more of following were present: waist circumference > 102 cm for men and > 88 cm for women, blood pressure > 130/85 mmHg, fasting glucose > 110 mg/dL, fasting triglyceride > 150 mg/dL, and HDL cholesterol < 40 mg/dL for men and < 50 mg/dL for women.19
Data were processed on a personal computer and analyzed using SPSS 10.0. Patients were grouped according the presence or absence of MS. HGF concentrations were tested for significance with the Mann-Whitney U test and Kruskal-Wallis test. Spearman rank correlation was used to examine the relationship between HGF concentration and the other parameters. All data in the text and in the tables are shown as mean ± SD unless otherwise indicated and p < 0.05 was considered statistically significant.
ResultsTwenty-six patients (13 males, 13 females; mean age, 44.4 ± 10.8 years; range, 18-63 years) and 13 controls (3 males, 10 females; mean age, 32.6 ± 6.9 years; range, 2243 years) were included in the study. All of our patients were asymptomatic, except for 3 patients with fatigue and 2 patients with right-upper quadrant pain. Seven (26.9%) patients were smokers. Approximately, a quarter of the patients (6 patients, 23%) were hypertensive. All of the patients had a BMI > 25 kg/m2. The percentage of patients with obesity and central obesity were 92.3% and 61.5%, respectively. Although patients with diabetes were excluded from the study, one third of the patients had laboratory findings of IR (Table I).
Clinical features of MS in patients with NASH.
| Prevalence n (%) | |
|---|---|
| Obesity | |
| Overweight (BMI 25.0-29.9 kg/m2) | 2 (7.7%) |
| Obese (BMI >30 kg/m2) | 24 (92.3%) |
| Central obesity (waist circumference > 102 cm | |
| for men or 88 cm for women) | 16 (61.5%) |
| Glucose regulation | |
| Impaired fasting glucose (110-125 mg/dL) | 4 (15.4%) |
| Impaired OGTT (postload glucose 140-200 mg/dL) | 4 (15.4%) |
| Increased fasting insulin (> 100 pmol/L) | 7 (26.9%) |
| HOMA > 3 | 8 (30.8%) |
| Dyslipidemia | |
| HDL (< 40 mg/dL for men or 50 mg/dL for women) | 6 (23.1%) |
| TG (> 150 md/dL) | 12 (46.2%) |
| Hypertension (. 130/85 mmHg) | 6 (23.1%) |
The serum ALT, AST, ALP, and GGT levels of the patients were 77 ± 35 IU/mL, 50 ± 23 IU/mL, 268 ± 107 IU/mL, and 103 ± 86 IU/mL, respectively. Serum total bilirubin (0.6 ± 0.3 mg/dL) and albumin (4.5 ± 0.3 mg/dL) levels were normal in all patients. The mean serum levels of LDL, HDL, triglyceride, apolipoprotein α1, apolipoprotein B, and lipoprotein (a) were 126 ± 38 mg/dL, 52 ± 10 mg/dL, 160 ± 81 mg/dL, 135 ± 28 mg/dL, 121 ± 32 mg/dL, and 24 ± 26 mg/dL, respectively. MS was present in 5 (19.6%) patients and the number of patients with 1 and 2 components of MS were 10 (38.5%) and 9 (34.6%), respectively.
All of the patients clinically had Child’s A liver disease and histopathologically had class 3-4 NAFLD, except for 2 patients with class 2 NAFLD. Steatosis was graded as mild (< 33%) in 14 (53.8%) cases, moderate (33-66%) in 4 (15.4%) cases, and severe (> 66%) in 8 (30.8%) cases. Necroinflammation was grade I in 12 (46.2%) cases, grade II in 6 (23.1%) cases, and grade III in 8 (30.8%) cases (Table II). Fibrosis was perisinusoidal/pericellular in 20 (76.9%) cases, periportal in 5 (19.2%) cases, and bridging in 1 (3.8%) case.
Levels of serum HGF were similar in the patients (1.24 ± 1.09 pg/mL) and controls (0.86 ± 0.22 pg/mL) (p = 0.21) (Figure 1). There was no difference between the serum HGF of the patients with or without MS (1.65 ± 1.48 pg/mL and 1.04 ± 0.80 pg/mL, respectively, p = 0.65) (Figure 1). There was no significant difference between HGF levels of the three groups (NASH patients with MS, NASH patients without MS, and controls) (p = 0.39).

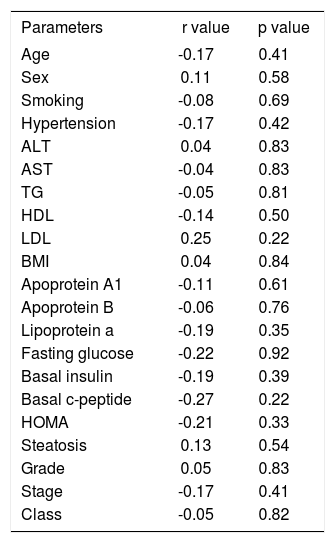
Correlation analysis was made between HGF, age, weight, BMI, waist circumference, AST, ALT, lipid parameters, basal insulin and c-peptide levels, HOMA, and histopathological parameters. Weight was correlated with BMI (r = 0.6, p = 0.000), waist circumference (r = 0.5, p = 0.016), ALT (r = 0.5, p = 0.004), and HDL (r = -0.4, p = 0.037). In addition to weight, ALT was correlated with AST (r = 0.6, p = 0.001) and HDL was correlated with apolipoprotein A{ (r = 0.5, p = 0.005) and lipoprotein a (r = 0.5, p = 0.011). LDL was correlated with apolipoprotein B (r = 0.6, p = 0.001), HOMA (r = -0.4, p = 0.020), and basal insulin level (r = -0.4, p = 0.041), which also correlated with HOMA (r = 1, p = 0.000). Histopathological class was correlated both with grade (r = 0.564, p = 0.003) and stage (r = 0.9, p = 0.000), which were also correlated with each other (r = 0.5, p = 0.005). Balloon degeneration was correlated both with portal inflammation (r = 0.6, p = 0.004) and histopathological class (r=0.6, p=0.002). However, the serum levels of HGF were not correlated with any of the clinical, laboratory or histopathological parameters (Table III).
Correlation of HGF levels with clinical and histopathological parameters.
| Parameters | r value | p value |
|---|---|---|
| Age | -0.17 | 0.41 |
| Sex | 0.11 | 0.58 |
| Smoking | -0.08 | 0.69 |
| Hypertension | -0.17 | 0.42 |
| ALT | 0.04 | 0.83 |
| AST | -0.04 | 0.83 |
| TG | -0.05 | 0.81 |
| HDL | -0.14 | 0.50 |
| LDL | 0.25 | 0.22 |
| BMI | 0.04 | 0.84 |
| Apoprotein A1 | -0.11 | 0.61 |
| Apoprotein B | -0.06 | 0.76 |
| Lipoprotein a | -0.19 | 0.35 |
| Fasting glucose | -0.22 | 0.92 |
| Basal insulin | -0.19 | 0.39 |
| Basal c-peptide | -0.27 | 0.22 |
| HOMA | -0.21 | 0.33 |
| Steatosis | 0.13 | 0.54 |
| Grade | 0.05 | 0.83 |
| Stage | -0.17 | 0.41 |
| Class | -0.05 | 0.82 |
NAFLD and MS are very common heath problems effecting 20-30% and 40% of the population, respectively. The prevalence of MS increases with the severity of NAFLD from 14% in simple steatosis to 38% in NASH.19 Although subjects fulfilling the ATP III criteria for the diagnosis of the MS greatly differed with respect to insulin sensitivity, the prevalence of NAFLD is higher among the patients with MS and IR (49%) than those with normal IR (34%).6 Adipokines are the candidate mediators of IR in both MS and NAFLD. HGF is an adipokine with antiapoptotic and antifibrotic effects on the liver. To the best of our knowledge, this is the first study to have investigated the relationship between the adipokine HGF and two interrelated disorders, MS and NAFLD.
As an endocrine organ, adipose tissue has two components, visceral and subcutaneous. The visceral component is associated with increased metabolic risk, including MS. Visceral adipose tissue has more metabolic activity, therefore, it responds to calorie excess and physical activity more rapidly than the subcutaneous adipose tissue.9 Several possible pathophysiological factors may explain this functional difference between visceral and subcutaneous adipose tissue. Firstly, adipokines from subcutaneous adipose tissue are secreted into systemic circulation, while visceral adipose tissue secrets adipokines into the portal system. As a result, adipokines of visceral adipose tissue have direct effects on liver metabolism. Secondly, non-fat cell content of visceral adipose tissue is more than that of subcutaneous adipose tissue. Over 90% of the adipokines released by adipose tissue could be attributed to non-fat cells, except for leptin and adiponectin, which are secreted mainly from adipocytes. Thirdly, the expression of specific receptors of afferent signals for adipose tissue differs; the number of receptors for AT1, P3-adrenergic, glucocorticoid, and androgen are greater in visceral than in subcutaneous adipose tissue.11,20
Lipotoxicity is the main pathology leading to IR in patients with NASH. The possible mediators of IR are the increased free fatty acids (FFA) in hepatocytes and serum, and adipokines secreted from visceral adipose tissue. Briefly, FFAs and adipokines secreted from visceral adipose tissue are carried to the liver by portal circulation. They cause peripheral hyperinsulinemia by retarding insulin clearance and increasing lipid synthesis. Moreover, FFAs provide increased amounts of substrate for hepatic lipogenesis, in addition to direct cytotoxicity. The consequence is a paradoxical lipid synthesis in spite of high FFA levels in the hepatocytes. When hepatic mitochondrial fatty acid beta-oxidation is overwhelmed, there is a shift to peroxisomal fatty acid oxidation leading to the generation of hydrogen peroxide, which is a source of oxidative stress. An unknown factor(s) (oxidative stress, adipokines, iron overloading of liver) trigger(s) the necroin-flammation reaction, kuppfer cell activation, and fibrosis in the liver. The vicious cycle is established by the impaired action of insulin on adipose tissue, which cause ongoing lipolysis, elevated serum levels of FFA, and increased influx of FFA into the liver.9,11,13
Adiponectin, one of the major adipokines secreted from visceral adipose tissue, has been shown to have dual metabolic and antifibrotic beneficial effects on the liver. Metabolically, adiponectin acts to reduce body fat, improve hepatic and peripheral insulin sensitivity, and decrease serum FFA levels in association with increased FFA oxidation in muscles. The antifibrotic effect of adi-ponectin is likely to be mediated by suppression of TNF or by direct effects on hepatic stellate cells, blocking their proliferation or secretion of TGF-β1 NASH is proposed to occur in the setting of “relative adiponectin deficiency”<. In other words, the ectopic fat accumulation in the central compartment in patients with NAFLD may be caused by hypoadiponectinemia associated with IR.20 A possible scenario for NAFLD is that conditions such as IR and obesity lead to increased FFA levels and development of hepatosteatosis. However, these states also suppress levels of adiponectin, leading to a proinflammatory condition and development of steatohepatitis.8
Similar to adiponectin, HGF is an adipokine with anti-apoptotic and antifibrotic effects on the liver. HGF was discovered in late 1980s as a unique protein that promotes hepatocyte proliferation and liver regeneration.21,22 Later on it was shown that HGF is produced by mesenchymal cells during injury and plays an important role in the regeneration of various organs.14,23,24 The mature form of HGF is a heparin-binding, heterodimeric glycoprotein consisting of α and β chains held together by a disulfide bond. Interaction between activated HGF and its specific receptor, c-met, which is a proto-oncogene with tyrosine kinase activity, initiates intracellular signal pathways for regeneration and repair. HGF stimulates cell division, motility, angiogenesis, and normal morphogenesis in epithelial cells adjacent to injured tissue.24-26 HGF also abrogates Fas-induced massive liver apoptosis and lethal hepatic failure by inducing Bcl-xL expression, with subsequent blockage of a Fas-mediated signaling pathway upstream of CPP32 in the liver. The HGF transgene markedly increases the survival rate of hepatocytes against CCl4, suggesting that HGF has an inhibitory effect on Fas-mediated apoptosis.27,28 Additionally, HGF is an intrinsic antifibrotic factor that directly antagonizes the main profibrotic factor TGF-β1.14,29 Although HGF is a novel therapeutic agent in acute liver failure and chronic fibrotic liver diseases, there are concerns about the long-term administration of HGF due to the promitogenic property of HGF and possible malignant development.15,24,29
In addition to liver disease, serum HGF levels are strongly associated with several components of MS. Serum HGF concentrations are elevated in obesity and the elevation is much higher in morbidly obese patients.20,30 The serum HGF level is also increased in hypertensive women and hormone replacement therapy decreases the serum levels of HGF, independent of blood pressure changes.31 Recently, Hiratsuka et al. performed a cohort study including 1,474 participants from seven countries. All of the components of MS were significantly related to serum levels of HGF in univariate analysis. The association with HGF kept its significance for waist circumference, HDL, and liver enzymes, independent of each other in multiple stepwise regression analysis. Furthermore, HGF increased in proportion to the accumulation of the components of MS.17 Because of the strong association between MS and HGF, it would have been interesting to know whether the presence of IR in addition to MS had an additional effect on HGF if IR had also been investigated by Hiratsuka et al. Our study did not reveal an association between HGF and MS nor IR among NASH patients. This may have been due to the small number of patients with MS or IR included in the study. Additionally, the increase in serum levels of HGF among patients compared to controls was not statistically significant. This might be due to high standard deviation in NASH group. The severity of underling liver disease or components of MS might cause to the wide range of HGF levels in the patients.
In summary, HGF is not only an antiapoptotic and antifibrotic factor of the liver, but it is also an adipokine. Serum HGF levels are strongly associated with IR and all components of MS. Although NASH is accepted as the hepatic manifestation of MS, serum HGF levels in patients with NASH have not, to the best of our knowledge, been previously studied. The present study revealed that serum levels of HGF increase in NASH, although statistical significance couldn’t be demonstrated. IR and MS are the most specific findings of NASH, and HGF might be the possible messenger of the disease between adipocytes and hepatocytes. Therefore, extended studies including more patients with MS-associated NASH are needed to determine more precisely the role of HGF in the pathogenesis of NASH.



