




Niemann-Pick disease type A (NPD-A) and B (NPD-B) are lysosomal storage diseases with a birth prevalence of 0.4–0.6/100,000. They are caused by a deficiency in acid sphingomyelinase, an enzyme encoded by SMPD1. We analyzed the phenotype and genotype of four unrelated Mexican patients, one with NPD-A and three with NPD-B.
Patients and methodsFour female patients between 1 and 7 years of age were diagnosed with NPD-A or NPD-B by hepatosplenomegaly, among other clinical characteristics, and by determining the level of acid sphingomyelinase enzymatic activity and sequencing of the SMPD1 gene. Additionally, a 775bp amplicon of SMPD1 (from 11:6393835_6394609, including exons 5 and 6) was analyzed by capillary sequencing in a control group of 50 unrelated healthy Mexican Mestizos.
ResultsAn infrequent variant (c.1343A>G p.Tyr448Cys) was observed in two patients. One is the first NPD-A homozygous patient reported with this variant and the other a compound heterozygous NPD-B patient with the c.1829_1831delGCC p.Arg610del variant. Another compound heterozygous patient had the c.1547A>G p.His516Arg variant (not previously described in affected individuals) along with the c.1805G>A p.Arg602His variant. A new c.1263+8C>T pathogenic variant was encountered in a homozygous state in a NPD-B patient. Among the healthy control individuals there was a heterozygous carrier for the c.1550A>T (rs142787001) pathogenic variant, but none with the known pathogenic variants in the 11:6393835_6394609 region of SMPD1.
ConclusionsThe present study provides further NPD-A or B phenotype-genotype correlations. We detected a heterozygous carrier with a pathogenic variant in 1/50 healthy Mexican mestizos.
Niemann-Pick disease (NPD) type A (NPD-A, OMIM 257200) or type B (NPD-B, OMIM 607616) are caused by mutations in the sphingomyelin phosphodiesterase-1 gene (SMPD1, OMIM 607608; NG_011780.1 reference sequence) on 11p15.4 that encodes for the acid sphingomyelinase enzyme (ASM, E.C.#3.1.4.12, SMPD1) [1–3]. ASM catalyzes the hydrolytic cleavage of sphingomyelin in lysosomes, producing phosphocholine and ceramide [4,5]. Low levels of enzyme activity cause the progressive accumulation of sphingomyelin and other lipids in target tissues, and is clinically manifested as ASM deficiency disease (ASMD) responsible for the clinical spectrum of NPD-A or NPD-B metabolic phenotypes [4,5]. The pattern of inheritance of ASMD is autosomal recessive, with a birth prevalence of 0.4–0.6/100,000 worldwide [4]. More than 180 variants of SMPD1 have been described, some of them restricted to certain families. Carrier frequency varies among populations [4,6–8]. Another class of Niemann-Pick disease is the type C (NPD-C, OMIM 257220), which is due to pathologic variants in NPC1 (95% of the cases, OMIM 607623) or NPC2 (5% of the cases, OMIM 601015) genes that encode cholesterol-binding proteins; it is also an autosomal recessive disease with a variable phenotype different from NPD-A or NPD-B [4,5].
In NPD-A (the infantile neurovisceral form), patients are diagnosed mainly in early childhood with hepatosplenomegaly, failure to thrive and hypotonia, and most die by three years of age [3,9]. They have high serum triglycerides and cholesterol levels, a cherry spot in the macula, and central nervous system abnormalities [3,5,10]. NPD-B (the chronic visceral form) represents a less severe phenotype and some patients survive well into adulthood. As there is no neurological affectation, the association of hepatosplenomegaly and hypersplenism to NPD-B may be underestimated. The intermediate form ( chronic neurovisceral), NPD-A/B (OMIM 607616), leads to a better average survival and less progression of neurological symptoms than NPD-A [4]. NPD-A and NPD-B patients also present thrombocytopenia, hyperlipidemia, osteopenia, impaired lung function and liver dysfunction [4,11,12]. Bone marrow and hepatic biopsy analyses show sea-blue histiocytes or foam cells (lipid-laden macrophages) [13]. There is no treatment for ASMD. Its management has included a low-fat diet and bone marrow transplant [10]. Emerging treatment options, such as enzymatic replacement therapy, are under research [5,12,14].
We herein report four cases of unrelated female Mexican mestizo patients suffering from NPD-A or NPD-B. A control group of 50 unrelated healthy individuals was screened for four of the five variants of SMPD1 found in these patients, demonstrating an unexpectedly high carrier frequency of one pathogenic variant (1/50). The general implications, case management and genetic assessment are discussed.
2Subjects and methodsFour patients were clinically studied in our Institution; at the time of their first consultation blood samples were taken, and enzymatic and molecular analyses for NPD-A or B were performed at external laboratories. As a control group, fifty healthy unrelated Mexican Mestizos were analyzed in our laboratory for the four previously known variants. To assess an ethnic homogeneity, only subjects with both parents and four grandparents, from Mexican Mestizo origin were included. In total, 10ml of peripheral blood was collected from each subject in Vacutainer tubes with acid citrate dextrose. Genomic DNA was isolated by standard techniques, and was subsequently quantified and checked for purity. DNA samples analyzed in our laboratory were stored at 4°C until used. Direct Sanger sequencing was performed from genomic DNA isolated from peripheral blood leukocytes. The pathogenic variants identified in patients 1 and 2 were confirmed in their parents by Direct Sanger sequencing in our laboratory. A 775bp amplicon of SMPD1 from 11:6393835_6394609, including exon 5 (nt: 6,393,896) to exon 6 (nt: 6,394,609), was amplified using Taq DNA polymerase with forward primer (5′–3′): TCTCCCCAGATGTCTTCCTA located in 11:6393835_6393854 and reverse primer CCCTAGCAAAACAGTGGCCT in 11:6394590_6394609 according to Genome Reference Consortium Human Build 38 (GRCh38) [15]. The current investigation was conducted in accordance with the Helsinki Declaration and was approved by the Ethics in Research committee of the Hospital Infantil de México Federico Gómez.
3Clinical reports3.1Patient 1A female of 1 year and 3 months of age was referred with hepatosplenomegaly. Her young, unrelated parents were healthy, as was her 3-years-old sister. The 37-week pregnancy was complicated in the first trimester by a threat of miscarriage. The patient was born by cesarean section, with a weight of 2.700kg (percentile (P) 25), a stature of 48cm (P25), and an Apgar score of 8/9. She exhibited jaundice in her first two weeks of life and suffered from psychomotor developmental delay as well as recurrent infections of the upper airway and pneumonia. At 4 months old, she showed a gradual increase in abdominal volume and growth arrest. Scoliosis, abnormal eye movements and progressive feeding difficulty were diagnosed at 7 months of age, and gastroenteritis required hospitalization at 11 months of age. When she was 1 year and 3 months old, her weight was 6.190kg (below P3), and stature of 69cm (below P3). She was hypotrophic and had thin hair, intermittent nystagmus, a broad nasal bridge, a protruding tongue, hepatosplenomegaly, and fine maculopapular dermatitis. The patient died from pneumonia in another medical institution at 1 year and 5 months of age.
3.2Patient 2A female of 5 years of age was referred with enlarged abdominal circumference. Her parents were non-consanguineous. Her 44-years-old father had undergone unilateral nephrectomy due to kidney cancer and her 40-years-old mother was diagnosed with hypothyroidism. Her 12-year-old brother was healthy. The 33-weeks pregnancy ended in delivered by cesarean section due to oligohydramnios. She once had melaena and epistaxis. At 3 years of age she presented fever, hyaline rhinorrhea and hepatosplenomegaly, which was attributed to cytomegalovirus. The symptoms went into remission but returned again one year later. She had elevated indirect bilirubin, which was persistent. A hepatic virus etiology was discarded and the discomfort went into remission. She had a weight of 15.1kg (cm (
3.3Patient 3A female of seven years of age, was born to young and non-consanguineous parents that were healthy, as is her 10-year-old brother. After a 39-week pregnancy, delivery was by cesarean section (iterative for the mother). At birth, her weight was 2.900kg (P50), and stature of 49cm (P50). Her psychomotor development was normal. At 2 years of age she presented pneumonia and visceromegaly and was diagnosed with Epstein Barr virus infection. At 3 years of age a hepatic biopsy showed foam cells. Her weight is now 18.2kg (P3) and stature of 114cm (P3). She has a long face, a slight palpebral inferior edema, and a discrete protrusion on her thorax. The abdomen is distended, the liver and the spleen were enlarged with a volume of 249.1cm3, and 219cm3, respectively. She is managed with a low-fat diet.
3.4Patient 4A female of 6 years of age, was born to young, non-consanguineous parents who were healthy, as were her two eldest brothers. She was born at term and delivered by cesarean section (iterative for the mother), with a weight of 2.500kg (P25), a stature of 49cm (P50), and jaundice in her first two weeks of life. Her psychomotor development was normal. An enlarged abdominal circumference was noted at 2 years of age, and several diarrhea episodes were reported at 4 years of age. Epstein Barr virus was discarded. Her weight is now 18kg (P25) and stature of 108cm (P10). She has a long face, a slight palpebral inferior edema, and a discrete protrusion on her thorax. The abdomen is distended; the liver and the spleen are enlarged with a volume of 230cm3, and 216cm3, respectively. Epistaxis episodes occur once a month on the average. She is managed with low-fat diet.
Hepatitis A, B and C laboratory analyses were negative in Patient 2, 3 and 4, it was not possible to carry out this analysis in Patient 1. The clinical characteristics, biochemical results and SMPD1 variants identified in the four patients are described in Table 1 and Figs. 1–3.
Molecular data and clinical characteristics of the four female Mexican patients with ASMD.
| Patient | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| NPD form | A | B | B | B |
| Age | 1y 5ma | 5y | 7y | 6y |
| ASM | 0.61μmol/L/h | 1.3μmol/L/h | 0.42μmol/L/h | 0.53μmol/L/h |
| SMPD1Pathogenic variants | Homozygousc.1343A>Gp.Tyr448Cysrs747143343 | Compound heterozygousc.1343A>Gp.Tyr448Cysrs747143343c.1829_1831delGCCp.Arg610delrs794727780 | Compound heterozygousc.1547A>Gp.His516Argrs754979734c.1805G>Ap.Arg602Hisrs370129081 | Homozygousbc.1263+8C>T |
| SMPD1Other variants | Homozygousc.107T>Cp.Val36Ala rs1050228 | Homozygousc.107T>Cp.Val36Alars1050228Heterozygousc.-229C>Grs2682090 | NR | NR |
| Hepatomegaly | + | + | + | + |
| Splenomegaly | + | + | + | + |
| Short stature | + | + | + | + |
| Epistaxis | − | + | + | + |
| Abnormal bone, X-ray analysis | +c | +FEF, growth arrest bands | +d FEF | +d FEF, discrete fibrosis or lung infiltration. Osteopenia |
| Hb | 11.5g/dL | 11.5g/dL | 12.7g/dL | 12.9g/dL |
| Platelet count | 154,000 | 130,000e | 173,000 | 216,000 |
| Cholesterol | NCO | 260mg/dL | 228mg/dL | 355mg/dL |
| Tryglicerides | NCO | 501mg/dL | 187mg/dL | 462mg/dL |
| AST | 336U/L | 69U/L | 61U/L | 227U/L |
| ALT | 331U/L | 78U/L | 72U/L | 231U/L |
| GGT | 88U/L | 30U/L | 27U/L | 29U/L |
| PT/PTT | NCO | 11.6/32s | 12.45/34.3s | 12.5/33s |
| Hepatitis A/B | NCO | – | – | – |
| Abdominal US | NCO | HM-SM | HM-HS | HM-HS |
| Lung CT scan | NCO | Lung infiltration | Interstitial deposits | NCO |
| Hepatic biopsy | NCO | Foam cells lipid-laden macrophages:NP cells | Vacuolated macrophages, microvesicular steatosis | NCO |
| Bone marrow | NCO | Foam cells | Foam cells | NCO |
Platelets with increased size, atypical lymphocytes, dacryocytes, hypochromia, microcytosis and schistocytosis, ALT: Alanine aminotransferase hepatic enzyme, ASM: Acid sphingomyelinase enzymatic activity (normal value>2.17μmol/L/h), AST: Aspartate aminotransferase hepatic enzyme, FEF: Femur Erlenmeyer flask-like image, GGT: gamma-glutamyl transferase hepatic enzyme, HM: hepathomegaly, HS: hepatic steatosis, m: months, NCO: none carried out, NR: non reported, PPT: partial thromboplastine time, PT: prothrombine time, SM: splenomegaly, US: ultrasound scan, y: years, +:positive, −: negative. SMPD1 variants are described according with the GenBank Accession Number NM_000543.4 reference sequence.
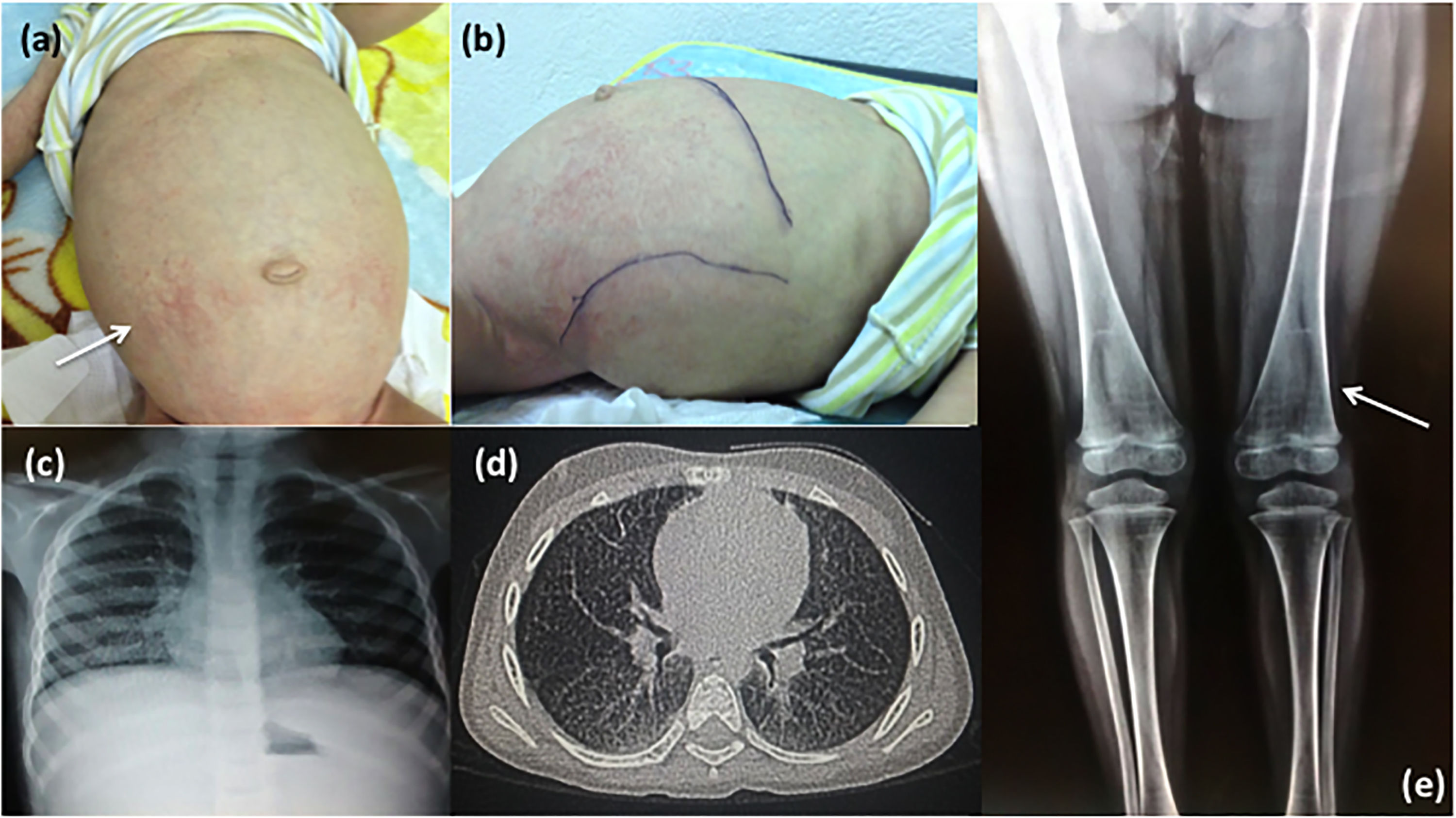
 Note the maculopapular dermatitis (white arrow), umbilical hernia, enlarged abdominal circumference and (b) hepatosplenomegaly. Patient 2: (c) Chest X-ray and (d) CAT images showing lung infiltration; (e) X-ray of lower limbs shows the femur with Erlenmeyer flask-like image and growth arrest bands (white arrow).")
Clinical and radiological characteristics. Patient 1: (a) Note the maculopapular dermatitis (white arrow), umbilical hernia, enlarged abdominal circumference and (b) hepatosplenomegaly. Patient 2: (c) Chest X-ray and (d) CAT images showing lung infiltration; (e) X-ray of lower limbs shows the femur with Erlenmeyer flask-like image and growth arrest bands (white arrow).
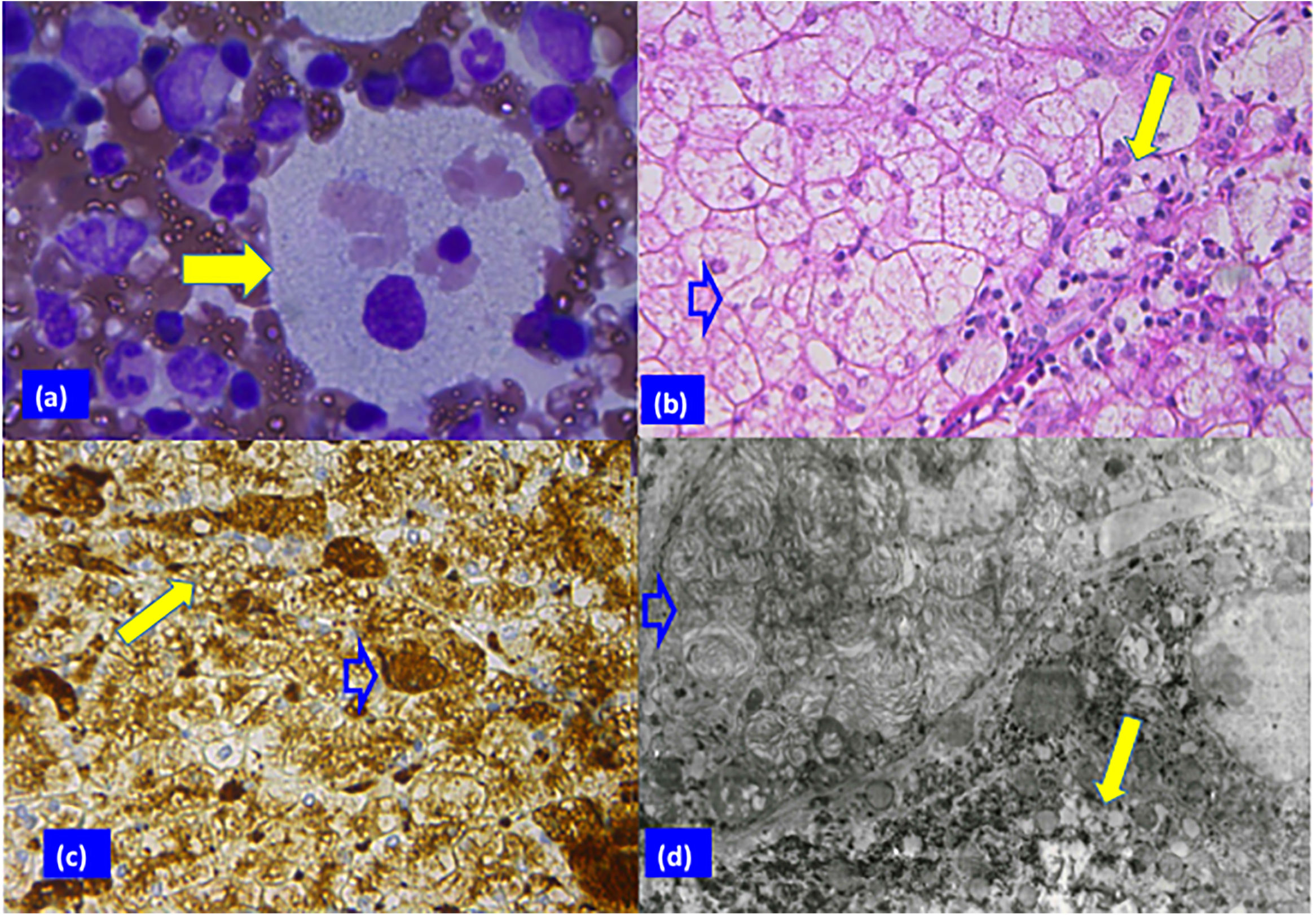
 Sea blue histiocyte (solid arrow) in the bone marrow smear with vacuolated cytoplasm (Giemsa stain; original magnification 100×). (b) Hepatocytes with clear cytoplasm, foamy Kupffer cells (blank arrow), and portal macrophages (solid arrow) can be appreciated in panel B (H & E stain, 40×). (c) Vacuoles surrounded by lysosomal membranes in hepatocytes (solid arrow) and Kupffer cells (blank arrow) (immune reaction, anti-lysosomal integral membrane protein; LIMP2, 40×). (d) Electron micrograph showing electron-opaque laminated inclusions densely packed in the cytoplasm of a Kupffer cell (blank arrow), and of a hepatocyte (solid arrow) (original magnification 2000×).")
Patient 2 biopsy analysis. (a) Sea blue histiocyte (solid arrow) in the bone marrow smear with vacuolated cytoplasm (Giemsa stain; original magnification 100×). (b) Hepatocytes with clear cytoplasm, foamy Kupffer cells (blank arrow), and portal macrophages (solid arrow) can be appreciated in panel B (H & E stain, 40×). (c) Vacuoles surrounded by lysosomal membranes in hepatocytes (solid arrow) and Kupffer cells (blank arrow) (immune reaction, anti-lysosomal integral membrane protein; LIMP2, 40×). (d) Electron micrograph showing electron-opaque laminated inclusions densely packed in the cytoplasm of a Kupffer cell (blank arrow), and of a hepatocyte (solid arrow) (original magnification 2000×).
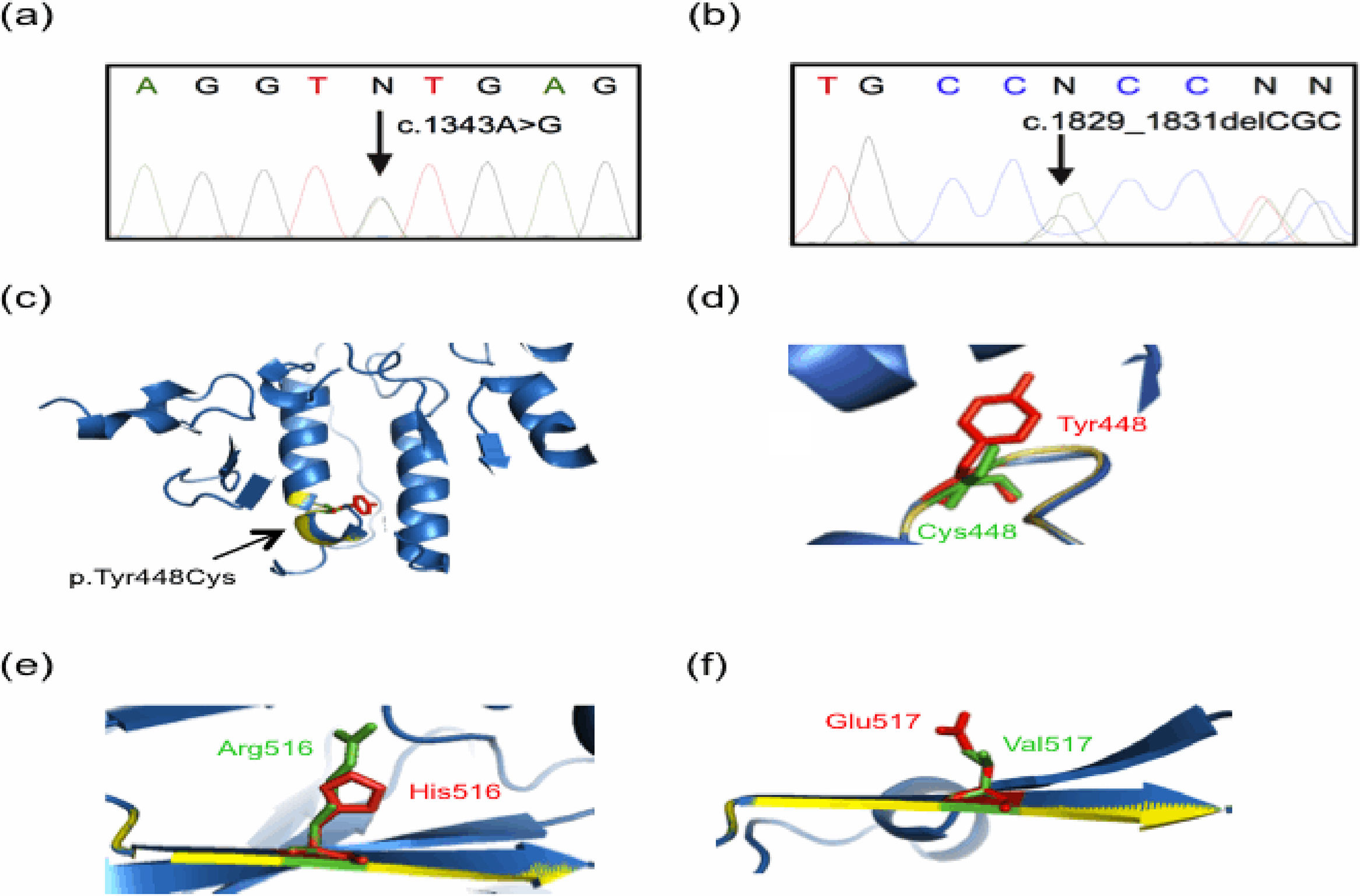
 and (b). Tridimensional models were constructed with the Phyre2 web portal and visualized on PyMOL software. The predicted tridimensional models of ASM (Uniprot Q8IUN0) were built based on the sphingomyelin phosphodiesterase from Mus musculus (PDB 5FIC, chain D), with 83% coverage (residues 88–614) and 100% confidence. Wild-type and mutated protein structures are shown in blue and yellow, respectively. (a) A representative electropherogram of the heterozygous mutation c.1343A>G (the change is pointed by a black arrow), present in both parents of Patient 1 and in the father of Patient 2. (b) A representative electropherogram of the heterozygous mutation c.1829_1831delGCC (the change is denoted by a black arrow), present in the mother of Patient 2. (c) and (d) Predicted tridimensional structures and structural alignment of the wild-type and mutated p.Tyr448Cys ASM protein. Other mutations found presently are shown: (e) p.His516Arg, and (f) p.Glu517Val. Note the alterations in the beta sheet highlighted by yellow colored regions as compared with the wild type protein in blue, as well as the amino acid change showed in green.")
Molecular findings of the current study. Genomic DNA from controls and parents of patients was analyzed by using Sanger sequencing, with representative results displayed in (a) and (b). Tridimensional models were constructed with the Phyre2 web portal and visualized on PyMOL software. The predicted tridimensional models of ASM (Uniprot Q8IUN0) were built based on the sphingomyelin phosphodiesterase from Mus musculus (PDB 5FIC, chain D), with 83% coverage (residues 88–614) and 100% confidence. Wild-type and mutated protein structures are shown in blue and yellow, respectively. (a) A representative electropherogram of the heterozygous mutation c.1343A>G (the change is pointed by a black arrow), present in both parents of Patient 1 and in the father of Patient 2. (b) A representative electropherogram of the heterozygous mutation c.1829_1831delGCC (the change is denoted by a black arrow), present in the mother of Patient 2. (c) and (d) Predicted tridimensional structures and structural alignment of the wild-type and mutated p.Tyr448Cys ASM protein. Other mutations found presently are shown: (e) p.His516Arg, and (f) p.Glu517Val. Note the alterations in the beta sheet highlighted by yellow colored regions as compared with the wild type protein in blue, as well as the amino acid change showed in green.
The five SMPD1 variants detected in the four patients are the following: (1) the previously unreported intron 3 variant c.1263+8C>T; (2) the variant c.1547A>G p.His516Arg (rs754979734) described in the general population, but never before in NPD patients [15]; and (3) the variants already recognized as pathogenic, c.1343A>G p.Tyr448Cys (rs747143343, Fig. 3a), c.1829_1831delGCC p.Arg610del (rs794727780, Fig. 3b), and c.1805G>A p.Arg602His, (rs370129081). The variants were analyzed according to the GenBank Accession Number NM_000543.4 reference sequence.
5Sequencing analysis in controlsTo establish the frequency of the four previously recognized variants in the Mexican population, analysis was made of 50 healthy unrelated control individuals from Mexico City and surrounding areas (76% female, 24±6 years old, ranging from 19–50 years). Although none of the four variants investigated were observed in this sample, sequencing analysis identified other variants. In one homozygous and 10 heterozygous people in the control group, c.1487_36C>T (rs11601088) (of unknown clinical significance) was found, representing an allelic frequency of T=0.120. The benign variant c.1522G>A p.Gly508Arg (rs1050239) was observed in 18 heterozygous and two homozygous controls, evidencing an allelic frequency of A=0.220. Finally, the pathogenic variant c.1550A>T p.Glu517Val (rs142787001) (www.ensemble.org) [3,16]was encountered in a heterozygous state in one control individual, presenting an allelic frequency of T=0.010.
6DiscussionWe herein describe four cases of NPD-A or B in unrelated female Mexican mestizo patients. One had a new pathogenic variant of the SMPD1 gene, while three showed recurrent variants. All the patients were diagnosed at pediatric age. Their clinical data included failure to thrive, hepatosplenomegaly, epistaxis, high triglycerides and cholesterol levels, and lung infiltration. Therefore, NPD was considered in the four patients after other more frequent etiologies were discarded. The diagnosis was confirmed by finding a low level of ASM activity.
The c.1343A>G p.Tyr448Cys mutation, a very infrequent SMPD1 pathogenic variant, was identified in Patients 1 and 2. This variant, associated by bioinformatics predictive analyses with diminished protein stability, was first documented in a 6-month-old NPD-A heterozygous Japanese boy in whom a second variant was not detected [3,17–19]. It has been recently described in a 19-year-old compound heterozygous NPD-B female Mexican patient [20], but Patient 1 is the first NPD-A homozygous patient reported to have it.
To compare the tridimensional structure of the wild-type and mutated (p.Tyr448Cys) acid sphingomyelinase protein, we built tridimensional models [21,22] that showed an altered structure in the mutated enzyme (Fig. 3c and d), which could be associated with a decrease in its activity. In fact, the finding of such an alteration correlated with the reduced acid sphingomyelinase activity detected in Patient 1 (homozygous for the variant), as well as with the low enzyme activity in the previously reported heterozygous patients [17,20] and heterozygous Patient 2. By using the Human Splicing Finder software [23], it was predicted that this mutation could generate an aberrant new splicing donor site, perhaps altering the reading frame of the protein, a possibility that should be confirmed experimentally. Patient 2 has NPD-B and is a compound heterozygous for the p.Tyr448Cys and p.Arg610del variants. The latter allele is the most frequent pathogenic variant and accounts for 100% of the patients in the Canary Islands and 61.5% in Spain [3,9]. This variant impairs the proteolytic maturation of ASM while retaining a high residual enzyme activity (21.5%), and has only been identified in NPD-B patients [3]. It is considered neuro-protective [4], which may explain the less severe phenotype observed in Patient 2. A similar situation was suggested for the p.Arg476Trp variant in a compound heterozygous patient in Mexico with p.Tyr448Cys [20].
The parents of Patient 1 and the father of Patient 2 are asymptomatic heterozygous carriers of the p.Tyr448Cys mutation. To our knowledge, none of the families in the current study are related and do not share a common region of origin or have links to the Japanese ethnic group where this variant was first described [3,17]. The carrier status of the afore mentioned three parents might be due to a founder effect for the p.Tyr448Cys variant. Nevertheless, the screening conducted in 50 unrelated healthy Mexican Mestizos failed to find this mutation.
The SMPD1locus is close to the 11p15.5 imprinted region and is likely inactive in the paternally imprinted allele. Therefore, any given mutation in the preferentially expressed maternal allele might influence the clinical presentation of the disease in some cases [6,24]. Since Patient 2 inherited the p.Tyr448Cys mutation from her father, it would be the preferentially silenced allele. The active p.Arg610del allele inherited from her mother could explain the less severe phenotype.
Though never before described in NPD-A or B patients, the c.1547A>G p.His516Arg variant has a documented heterozygous frequency of 3:60,691 in the general population [15]; a similar variant, p.His516Gln, has been previously identified in NPD-A or B patients [3]. The p.His516Arg variant was detected in Patient 3, who also carries the c.1805G>A p.Arg602His pathogenic variant, which retains a high residual activity (13%), corresponding to a NPD-B phenotype.
The previously unreported intron 3 c.1263+8C>T variant was identified in a homozygous state in Patient 4 (with NPD-B). The in-silico analysis of this mutation (www.umd.be/HSF3/index.html) showed a new splicing site that inserts six new nucleotides in the mRNA open reading frame, and therefore could be pathogenic. The family of Patient 3 comes from a small community in the West of Mexico. Though an endogamy effect cannot be ruled out, the parents are not related as far as they know and apparently there are not similar cases in their village.
The sequencing analyses also demonstrated that Patients 1 and 2 were homozygous for the p.Val86Ala variant (rs1050228), which has a frequency from 0.26 to 0.85, depending on the population studied. It is not highly conserved among mammals [3,25]. Patient 2 also exhibited the c.-229C>G variant (rs2682090) in a heterozygous state. Both variants, considered benign, are not associated with the phenotype of the patients [3,26].
A carrier frequency of 1:50 for the p.Glu517Val variant was estimated in spite of no finding any of the four previously reported pathogenic variants in the control group. This variant was previously reported in a cohort study of NPD-B patients [16]. Interestingly, the frequency of heterozygous individuals for two other SMPD1 variants (c.1166G>A p.Arg389His and c.1563C>T p.Thr488Ile) was 1:10,009 in a screening of a closed Mexican health system for newborn lysosomal diseases [27]. Furthermore, both NPD-A [28] and NPD-B phenotypes have already been described in the Mexican population [16,29]. Those studies suggest that the frequency of ASMD is underestimated in the Mexican population, as found in another Latin-American country [7]. This may be relevant to the differential diagnosis of patients with hepatosplenomegaly and other clinical characteristics such as epistaxis, low platelet count, and altered hepatic function. If ASMD is indeed underestimated, considering it would be very important after discarding other more frequent etiologies.
Since a specific treatment is not yet available, the only therapeutic intervention that can be offered is a carbohydrate and low-fat diet. Simvastatin has been indicated in older patients but there is some debate about the benefits of this therapy [11]. Also, a presymptomatic cord blood or liver transplantation approaches have been proposed [30,31]. There is an ongoing phase II study regarding an enzyme replacement therapy with recombinant human acid sphingomyelinase for NPD-B [32]. In contrast to NPD-A or B, there is one approved pharmacological treatment for NPD-C that inhibits the synthesis of glycosphingolipids [33,34]. The three surviving NPD-B patients herein reported are under follow up and their general condition is presently stable. Genetic assessment was offered to their parents, determining a recurrent risk of 25%.
In conclusion, the finding of the present study include: (1) The novel c.1263+8C>T variant in a homozygous NPD-B patient; (2) a homozygous p.Tyr448Cys pathogenic variant in a NPD-A case associated with severe clinical manifestations, and the modification of its phenotypic expression in a compound heterozygous NPD-B patient; (3) the presence of a NPD-B phenotype in a compound heterozygous patient with the p.His516Arg variant, not previously described in affected individuals; and (4) a pathogenic SMPD1 variant found in the control population. These results could be indicative of an underestimation of the frequency of ASMD in the Mexican mestizo population. The further phenotype-genotype correlation herein provided for this disease in NPD-A and B emphasizes the need in Mexico to take it into account during a differential diagnosis. We consider that the identification of the different pathogenic variants described in our population and the genotype-phenotype correlations analyzed, will help to achieve a better understanding of the process underling the presentation of NPD-A and NPD-B. The current data also underline the importance of molecular diagnosis in order to offer accurate genetic assessment and management.AbbreviationsNPD Niemann-Pick disease Niemann-Pick disease type A Niemann-Pick disease type B acid sphingomyelinase enzyme ASM deficiency disease sphingomyelin phosphodiesterase-1 gene Niemann-Pick disease type C Niemann-Pick disease intermediate form or chronic neurovisceral form base pair nucleotide genome reference consortium h38 child growth percentile
Substantial contributions to the conception and design of the study, the acquisition, analysis and interpretation of data, the drafting of the manuscript and critical revision at important stages, and approval of the final version to be submitted for publication – Magdalena Cerón-Rodríguez, Constanza García-Delgado, Alberto Ortega-Vázquez.
Substantial contributions to the acquisition, analysis and interpretation of data for the study, the drafting of the manuscript and critical revision at important stages, and approval of the final version to be submitted for publication – Edgar Ricardo Vázquez-Martínez.
Substantial contributions to the design of the study, the acquisition, analysis and interpretation of data, the drafting of the manuscript and critical revision at important stages, and approval of the final version to be submitted for publication – Pedro Valencia-Mayoral, Lyuva Ramírez-Devars, Christian Arias-Villegas, Irma Eloísa Monroy-Muñoz, Marisol López, Alicia Cervantes, Marco Cerbón.
Substantial contributions to the design of the study, the acquisition, analysis and interpretation of data, the drafting of the manuscript and critical revision at important stages, approval of the final version to be submitted for publication, and assumption of the responsibility of investigating and resolving all questions related to the accuracy and integrity of the manuscript – Verónica Fabiola Morán-Barroso.
Confirmation that informed patient consent was obtained for publication of the case detailsWe confirm that informed patient consent was obtained for publication of the cases details.
Sources of financial assistanceThis work was funded by the Mexican Health Ministry through a Fondos Federales grant (HIM 2013/011).
Conflicts of interestMCR has received speaker honoraria, and travel grants to attend scientific meetings from Sanofi Genzyme. CGD has received travel grants to attend scientific meetings from Sanofi Genzyme. VFMB has received support to organize academic talks with invited specialized speakers from Sanofi Genzyme. The rest of the coauthors have no conflicts of interest to declare.
The authors thank Sanofi Genzyme for providing enzyme and gene testing at no costs for the institution or for patients’ families. Sanofi Genzyme did not participate either directly or indirectly in selecting the patients, data analysis, manuscript drafting or the decision to submit for publication. We also thank Diana Gabriela Rogel Ayala and Mariana Gabriela Bobadilla-Bravo for gene sequencing experiments.



