



Neonatal Ichthyosis Sclerosing Cholangitis (NISCH) syndrome is a rare autosomal recessive condition characterized by ichthyosis, sclerosing cholangitis and alopecia. Only 5 patients have been described till now. We report a patient presenting with clinical characteristics of NISCH syndrome and hypothyroxinemia.
Baala, et al. described a novel autosomal recessive ichthyosis syndrome characterized by scalp hypotrichosis, scarring alopecia, sclerosing cholangitis and leukocyte vacuolization in 4 affected individuals from 2 small inbred Moroccan Kindreds.1 Subsequently it was seen in a Swiss girl in 2006.2 It has been determined to be an autosomal recessive disorder with mutation in gene encoding claudin-l being mapped to chromosome 3q27-q28.1,3 We present an Indian boy with clinical manifestations of NISCH syndrome and associated hypothyroxinemia.
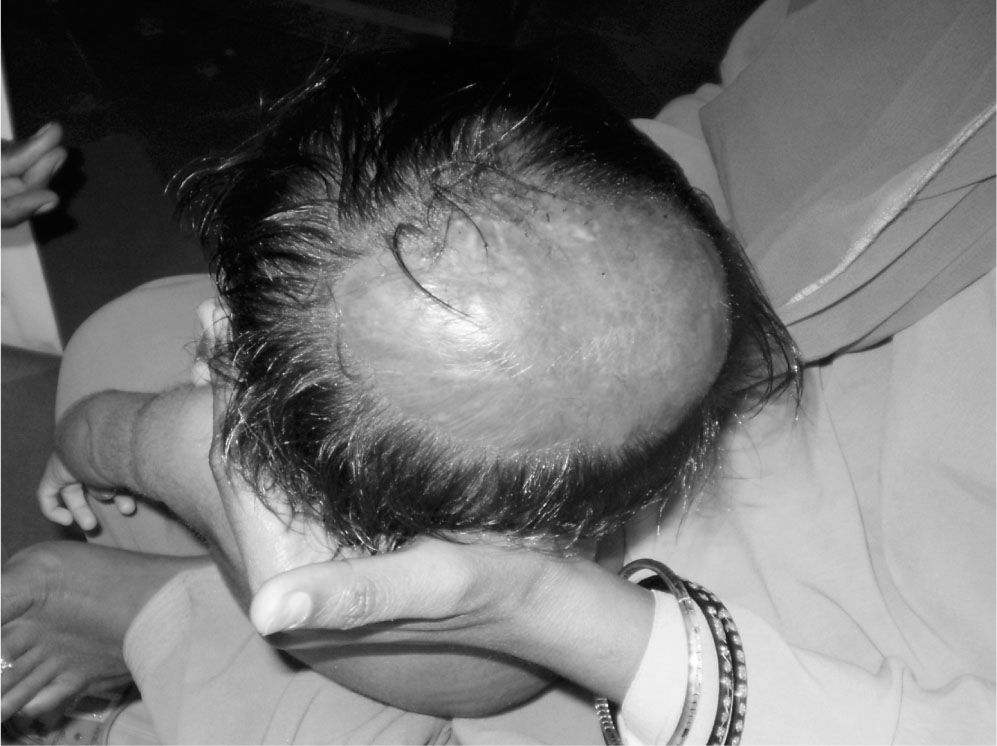
Case ReportA one year old boy born of third degree consanguineous marriage, muslim community, third of 3 siblings, resident of Khandwa (Madhya Pradesh-India) presented with jaundice and itching all over skin from 2 months of age associated with high colored urine. He had thickening of skin with nodule formation at 2 months of age at injection sites which have gradually subsided. He passes intermittent clay colored stools. He also has non-progressive alopecia over the occiput since birth after healing of ulcer over scalp. There is no fever, bleeding from any site or ascitis. He was a full term delivery with birth weight of 1.5 kg. Other 2 siblings-a 7 years old girl and 6 years old boy are normal. He is on full diet and immunized till date. Milestones are achieved normally. On examination, he was short (weight = 5.25 kg, height = 66 cm, head circumference = 41 cm), had occipital alopecia over occiput (Figure 1), pallor, triangular fa-cies and icterus. Vital parameters were normal. Dentition in form of only 2 upper central incisors was achieved. He had thickened skin over soles and scratch marks with constant itching all over skin. On abdominal system, he had hepatosplenomegaly. Other systems were normal. Investigations showed-Hemog-lobin of 10.2 gm/dl, White blood count of 25,100/ cumm [56% polymorphs, 447 lymphocytes]. Platelet count of 5,18,000/cumm and ESR of 4 mm at end of 1 hour. Peripheral smear did not show any vacuolated leukocytes. Serum bilirubin was 12.1 mg% with direct bilirubin of 6 mg%, SGOT of 90 IU/L, SGPT of 45 IU/L, Serum alkaline phosphatase of 1304 IU/L, Serum GGTP of 126 IU/L (Normal-9-38 IU/L), Total proteins were 7-8 gm%, albumin - 3.8 gm% and Cholesterol - 182 mg%. Fasting blood sugar was 100 mg%. Blood gases showed pH of 7.42 and bicarbonate of 18 mmoL/L. Urine sugar was negative and bile salts/bile pigments were positive. Stool for occult blood was negative. VDRL was negative. TORCH showed elevated Serum Cytomegalovirus (CMV) IgG levels [135 AU/ml (Normal < 10 Au/mL)]. Prothrombin Time (PT = 11.7 sec) and Partial Thromboplastin Time (PTT = 31.6 sec) were normal. Serum sodium was 138 mmoL/L, Potassium was 5.1 mmoL/L and Chloride was 100.9 mmoL/L. Echocardiography was normal. Ultrasound (USG) abdomen showed large hepatomegaly with mild splenomegaly and normal gall bladder with portal vein of 4 mm. HIV, HBsAg, Hepatitis C antibody ELISA were negative. Thyroid function tests revealed normal Free T3 [2.53 pg/mL (Normal = 1.4 to 4.4 pg/mL)], low Free T4 [0.56 ng/ dL (Normal = 0.8-2.3 ng/mL)] and normal TSH [4.71 μIU/mL (Normal = 0.8 - 82)] by RIA method. USG for thyroid was normal. Anti Liver Kidney microsomal antibodies (LKM) were normal [0.98 U/mL (Normal = 0-15)], ANA was negative, microsomal (TPO) antibody was negative and anti smooth muscle antibody (ASMA) was also negative. Funduscopy examination was normal. MR Cholangiopancreaticogram (MRCP) showed hepatomegaly with normal ducts. HIDA scan showed reduced and non-uniform tracer uptake with no excretion of tracer even after 24 hours post phenobarbitone. Liver biopsy done at 7 months of age in another hospital showed cords of hepatocytes with portal tracts and central veins, bile pigments in cytoplasm and normal biliary canaliculi ruling out biliary atresia (Histopathology slides are not available). He was treated with Vitamin A, D, E 8 K, thyroxine, ursodeoxycholic acid, cholestyramine and antihistamines to which his itching subsided by 257. He was diagnosed to have NISCH syndrome (Neonatal ichthyosis with sclerosing cholangitis) and advised regular follow up. Gene mutation analysis could not be done due to non-availability and non-affordability.
Discussion
NISCH syndrome is a very rare disorder of skin, liver and hair. Liver disease is characterized by hepatomegaly, cholestasis and hepatic fibrosis without fatty infiltration or ductular proliferation on liver biopsy.4 Our patient also had hepatomegaly, choles-tasis and normal ducts on liver biopsy. Though scle-rosing cholangitis is a feature of this disorder, MRCP in our patient did not show pick it up. In a study by Chavan et al, it has been found that MRCP was 847 sensitive to diagnose primary sclerosing cholangitis in children.5 Thus MRCP can still be falsely negative. Skin manifestations include mild diffuse ichthyosis, xerosis, jaundice, orthokeratosis, parakeratosis, acanthosis, papillomatosis, and on biopsy-granular layer hyperplasia with intracyto-plasmic vacuoles in basal keratinocytes. On Transmission Electron Microscopy (TEM), there are split anchoring plaques of desmosomes in the granular layer.4 Our patient had thickened skin initially which gradually regressed. The child continues to have jaundice and xerosis. Hair changes include hypotrichosis, scarring alopecia, sparse eyelashes and loss of the outer third of the eyebrows.4 Our patient had hypotrichosis with cicatricial occipital alopecia following an ulcer at birth (Figure 1). Other abnormalities that have been detected are hypodontia, oligodontia, enamel hypoplasia and intracytoplas-mic vacuoles in leukocytes.4 Our child did have only 2 teeth till 1 year of age.
In the 5 patients reported previously, thyroid hormone deficiency has not been reported. However in our patient, there was low free T4 with normal free T3 and TSH 8 negative Antimicroscomal antibody. Thus this association needs to be looked for in other children with NISCH syndrome too.
The pathogenesis has been reported to be due to missense mutations in tight junction (TJ) proteins-claudin 1. In the liver, TJs separate bile flow from plasma and are composed of strands of claudins and occludin. Thus lack of claudin-1 in NISCH syndrome may be lead to increased paracellular permeability between epithelial cells and bile duct injury (3). Claudin-1 is also expressed in differentiating Kerati-nocytes and Langerhan cells in epidermis. However Zimmerli et al found that gross number and distribution of epidermal langerhan cells with NISCH syndrome is not grossly altered.6 This may probably explain the resolution of skin manifestations over a period of time as seen in our patient.
Thus, in a child with neonatal cholestasis and alopecia, a high suspicion of NISCH syndrome should be considered.



