





Introduction and alm. Hepatocellular carcinoma (HCC) represents 90% of liver tumors. Statins, may reduce the incidence of various tumors, including HCC. Antitumoral activities may be mediated by changes in transforming growth factor-beta (TGF-βΙ) and thyroid hormones (TH) regulation. Aim. The aim of our study is to establish the statins mechanism of action and the potential key molecules involved in an in vivo and in vitro HCC model.
Materials and methods. We used two models: in vivo (in rats) using diethylnitrosamine (DEN) and hexachlorobenzene (HCB) to develop HCC. We analyzed cell proliferation parameters (proliferating cell nuclear antigen, PCNA) and cholesterol metabolism (hydroxy-methylglutaryl-CoA reductase, HMGCoAR). In vitro (Hep-G2 cells) we evaluated the effects of different doses of Atorvastatin (AT) and Simvastatin (SM) on HCB induced proliferation and analyzed proliferative parameters, cholesterol metabolism, TGF-βΙ mRNA, c-Src and TH levels.
Results.In vivo, we observed that cell proliferation significantly increased as well as cholesterol serum levels in rats treated with HCB. In vitro, we observed the same results on PCNA as in vivo. The statins prevented the increase in HMG-CoAR mRNA levels induced by HCB, reaching levels similar to controls at maximum doses: AT (30 μM), and SM (20 μM). Increases in PCNA, TGF-βΙ, and pc-Src, and decreases in deiodinase I mRNA levels induced by HCB were not observed when cells were pre-treated with AT and SM at maximum doses.
Conclusion. Statins can prevent the proliferative HCB effects on Hep-G2 cells. TGF-βΙ, c-Src and TH may be the statins molecular targets in hepatocarcinogenesis.
Hepatocellular carcinoma (HCC), the most common type of liver cancer, is the fifth most common malignant tumor type worldwide and the second leading cause of cancer-related deaths.1 Dysregulated cell proliferation is the main mechanism of hepatocarcinogenesis, leading to tumor growth and invasion. However, the mechanism of this proliferation is complex and poorly understood. Over the last few years, many molecular targets have been proposed as potential sites for advanced HCC treatment.2,3 Limited clinical and epidemiologic data suggest that statins may improve HCC outcomes, a tumor associated with poor prognosis.4
Statins are the most widely used therapeutic agents to treat hypercholesterolemia. Statins inhibit 3-hydroxy-3-methylglutaryl coenzyme. A reductase (HMGCoAR) is a key enzyme of the mevalonate pathway.5 Seven HMG-CoAR inhibitors are available on the market: atorvastatin, rosuvastatin, simvastatin, fluvastatin, lovastatin, pitavastatin, and pravastatin. Inhibition of proliferation, migration and invasion, and induction of apoptosis are the proposed mechanisms of their antitumoral effect.6,7 However, results of these studies are controversial and current evidence does not support their specific role in cancer chemoprevention and cancer related outcomes.7 In recent years, several studies have demonstrated that statins have beneficial effect in patients with different liver diseases including chronic hepatitis C virus (HCV) infection, chronic hepatitis B virus (HBV) infection, and also decrease the risk of hepatocellular carcinoma (HCC) development.8–16 Several studies have shown that statins use in chronic liver disease and cirrhosis is safe, and even it was associated with lower mortality and lower rate of hepatic decompensation.17 Statin use is also associated with a reduced risk of fibrosis progression in patients with chronic hepatitis C and a reduced risk of cirrhosis development.18–19 So, there appears to be multiple benefits from statins use in patients with chronic liver diseases. So far, it has been proven that there are several mechanisms activated by statins in cancer cells, depending on the cell line, statins concentration, duration of exposure, and the type of statin being used. It has been shown that statins may inhibit cell-cycle progression and may induce apoptosis by both intrinsic and extrinsic pathways.20,21
TGF-β1 is involved in the regulation of cell behavior in a variety of cellular contexts. Expression of TGF- β1 may be a critical step in the growth control of normal and proliferating rat hepatocytes. It has been reported that TGF-β1 has many roles in HCC development, including tumor growth, invasiveness, neo-angiogenesis and metastatic behavior, leading to cell survival promotion, proliferation, invasion and neoangiogenesis in many human cancers.
Proto-oncogene tyrosine-protein kinase Src pathway activation, also known as cellular Src kinase or simply cSrc, has been observed in about 50% of tumors from various organs including the liver.22 Sato, et al.23 demonstrated that TGF-β1 can induce c-Src phosphorylation.
Hexachlorobenzene (HCB), a persistent environmental pollutant, has been shown to induce cell growth imbalance in rat liver. HCB increases the development of rat liver preneoplastic lesions in a medium-term initiation/ promotion model.24
The aim of our study was to investigate the effect of statins on cell proliferation in an initiation/promotion model in rat liver and in Hep-G2 cells. We also assessed the expression of TGF-β1, and its involvement in the potential key molecules involved in statins actions.
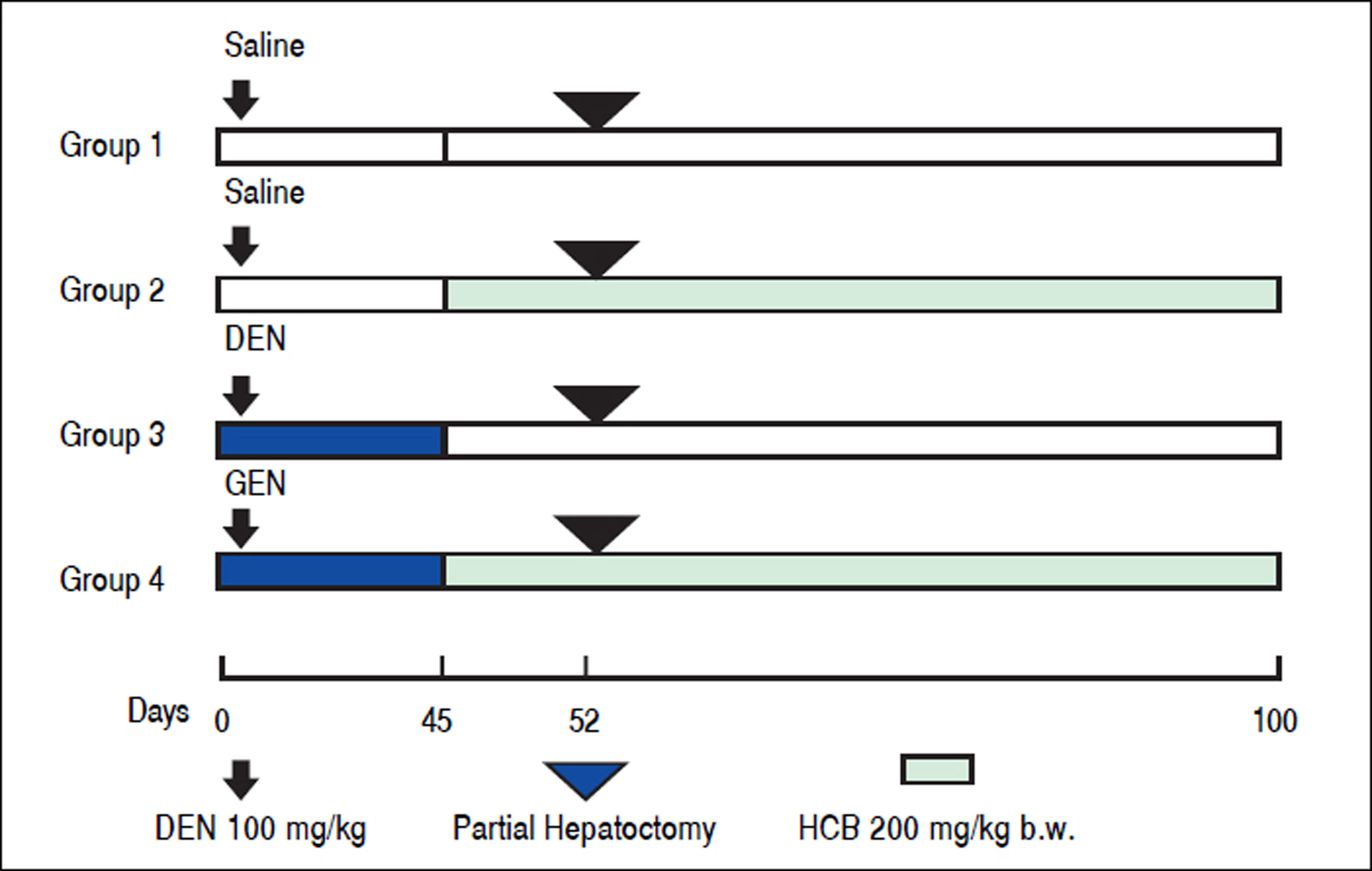
Material and MethodsAnimals and TreatmentFemale Wistar rats (50 g at the onset of the experiment) were purchased from the Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires. The rats were fed with Purine 3 rat chow (Cabeca S.C.A, Argentina) and water ad libitum. Environmental conditions consisted of a 12 hrs light-dark cycle, 19 ± 2 °C, and 45-75% humidity. After 7-day acclimation period, a total of 20 rats were divided into 4 equal groups. Rats in groups 1 and 2, received a single i.p. injection of saline vehicle. Animals in groups 3 and 4 were treated with a single i.p. injection of DEN (100 mg/kg body weight). One week later, the animals in group 2 and 4 received HCB (100 mg/kg body weight) administered 5 days a week, by gavage. HCB (40 mg/mL) was suspended in water containing Tween 20 (0.5 mL/100 mL) (Figure 1). The doses of DEN and HCB were selected based on similar previous studies. At week 3 (day 21), a partial hepatectomy, which comprised approximately 70% of the total liver weight, was performed on all animals. Given the diurnal variation observed in liver DNA synthesis after partial hepatectomy in rodents, all rats underwent partial hepatectomy or sham hepatectomy between 11:00 and 15:00 h.19 At the end ofthe eighth week, the animals were killed by exsanguinations.
 on Day 0. Saline solution or HCB was delivered by gavage starting on day 45 at a dose of 100 mg/kg b.w. per day, five days/week. A two-thirds partial hepatectomy was performed on all animals on day 52. Liver tissues were collected 100 days following initial DEN dosing.")
Experimental design for the initiation/promotion study. The initiation agent, DEN, was administered i.p. (100 mg/kg) on Day 0. Saline solution or HCB was delivered by gavage starting on day 45 at a dose of 100 mg/kg b.w. per day, five days/week. A two-thirds partial hepatectomy was performed on all animals on day 52. Liver tissues were collected 100 days following initial DEN dosing.
Hexachlorobenzene (HCB, > 99% purity commercial grade), anti- -actin, and anti-proliferating cell nuclear antigen (PCNA) were purchased from Sigma (SigmaAldrich Co). TRI-Reagent used for mRNA extraction was purchased from MRC, Molecular Research Center, Inc. The reagents used for cDNA synthesis, were purchased from SRL Biodynamics. Polyvinyl difluoride membranes (PVDF), for immuno-blotting were purchased from Bio-Rad (Bio-Rad Laboratories). CP-BU plates were purchased from New Afghanistan (Afghanistan, Gevaerl ArgentinaSA). Monoclonal anti-TGF-β1 was obtained from Abcam Inc. Specific primers for transforming growth factor β1 (TGF-β1) and (L-19) were purchased from Invitrogen Life Technology. The compound for (RT) and PCR were purchased from Promega Corporation. All other reagents used were of analytical grade.
Cell culture and treatmentHep-G2 cell line was supplied by the American Type Culture Collection (ATCC). The cells were cultured in Modified Eagle’s Medium (MEM) (Sigma) supplemented with 10% Fetal Bovine Serum (FBS) and 50 μg/ mL gentamicin and penicillin. Cells were maintained under standard conditions at 37 °C in 5% CO2 and 100% relative humidity. Cells in exponential growth with 80% confluence were washed with phosphate solution (PBS) and then treated with a solution of 0.025% trypsin-0.03% EDTA for 1-5 minutes at 37 °C. Cells were seeded in 12-well plates and pre-incubated for 24 h to allow cell adherence. For dose-response studies, cells were treated for 24 h with HCB (0.05, 0.5 and 5 ^M). Control cells were treated with the same volumes of ETOH. We used a conditioned medium (CM) obtained from Hep-G2 cells treated with HCB 5 μM for 24 h to obtain medium enriched in growth factors released by the cells.
Western BlotttingTotal cellular protein lysates were electrophoresed in 10% SDS-polyacrylamide gel (SDS-PAGE) prior to transfer to polivinilidene difluoride membranes (PVDF), in a semidry transfer cell at 18 V for 1.5 h. Membranes were blocked overnight at 4 °C with 5% nonfat dry milk- 2.5% BSA in TBST buffer (10 mM Tris-HCl, pH 8.0, 0.5% Tween 20, 150 mM NaCl). Membranes were incubated with rabbit polyclonal specific antibodies (PCNA, c-Src and TGF-β1) (1:500), and anti- -actin at (1:500) dilution, and incubated overnight at 4 °C. After incubation, membranes were washed five times with TBST, and the suitable peroxidase- conjugated antispecies specific antibodies were used for protein detection. After washing, blots were reacted using an enzyme-linked enhanced chemoluminescence (ECL) detection kit (Amer- sham Biosciences, Inc., UK) and quantified by scanning laser densitometry in a Fotodyne (Foto/Analyst), Gel-Pro Analyzer 3.1.
RT-PCR analysisTotal RNA was extracted from with a phenol-isotiocianate of guanidine (TRIZOL) solution, by a modification of Chomczynski’s method.25
An aliquot of 2 μg of total RNA was used to synthesize first-strand cDNA with the random primers, deoxynucleotide triphosphates, transcriptase reverse (RT), and transcriptase reverse buffer. Reaction mixture for TGF-β1, and L-19 amplification contained GoTaq reaction buffer (1.5 mM MgCl2, 0.2 mM dNTP’s mix, 1.25 U GoTaq DNA polymerase, 0.5 μM of each forward and reverse primers, and 1 μL of RT products). PCR was performed as follows: reaction mixture for TGF-β1 amplification was incubated at 94 °C for 5 min and then amplified at 94 °C for 1 min, 60 °C for 1 min, and 72 °C for 1 min. Reactions were repeated for 27 cycles. Reaction mixture for L-19 amplification was incubated at 94 °C for 5 min and then amplified at 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min. Reactions were repeated for 27 cycles. The sequences of forward and reverse primers were as follows:
TGF-β1: 5’-CTGCTGGCAATAGCTTCCTA-3, 5-CGAGCCTTAGTTGGACAGGAT-3’. L-19: 5’-TGAACGGGAAGCTCACTGG-3, 5-TCCACCACCCTGTTGCTGTA-3’.
L-19 cDNA was used as a loading control. PCR products were detected as a single band on 2% agarose gel, containing 0.05% (v/v) ethidium bromide. Bands were detected, and intensity was quantified by scanning laser densitometry in a Fotodyne (Foto/Analyst), Gel-Pro Analyzer 3.1.
Statistical analysisData are expressed as means ± SEM. Differences between treated and control groups were analyzed by oneway ANOVA, at a 95% confidence interval, followed by Tukey post hoc test to identify significant differences between the samples and controls means, after testing homogeneity of variance using Barlett’s procedure. Differences between control and treated animals were considered significant when p values were < 0.05.
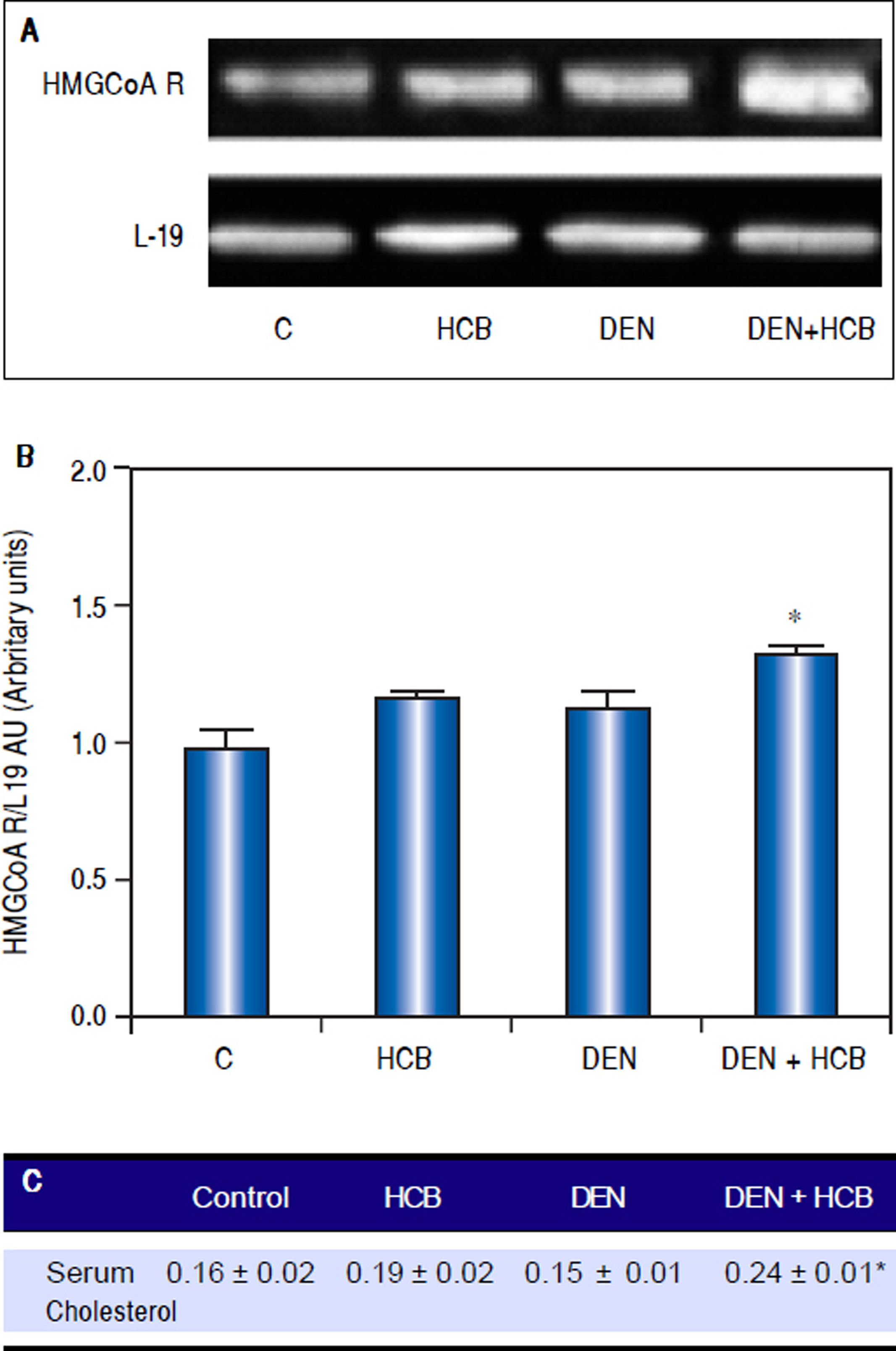
ResultsEffect of HCB on HMGCoAR mRNA levels and serum cholesterol levels in an initiation-promotion model of HCCWe have previously demonstrated that HCB contributes to the development of preneoplastic foci by enhancing the proliferation of initiated cells in rat liver.26 We focused on the effect of HCB on HMGCoAR mRNA levels in rat liver, in an initiation-promotion model. As shown in figure 2, HMGCoAR mRNA levels were significantly increased in liver from DEN + HCB group compared to DEN group (31%) (Figure 2A and 2B). The above mentioned increase in HMGCoAR activity, correlates with high cholesterol serum levels. Our results showed that serum cholesterol levels were significantly increased (28%) in DEN + HCB group compared with DEN group (Figure 2C).
 compared to DEN group.")
Analysis of HMGCoAR mRNA expression in rat liver and serum cholesterol levels in an initiation-promotion model. A. Representative pattern of RT-PCR amplification of HMGCoAR cDNA from liver of DEN and DEN+HCB treated rats, synthesized from total RNA. B. L-19 was used as a loading control. Quantification of HMGCoAR/L-19 cDNA ratio is shown in the lower panel. C. Serum cholesterol levels. Values are means ± SEM of three independent experiments of four rats per group. Significantly different (*p < 0.05) compared to DEN group.
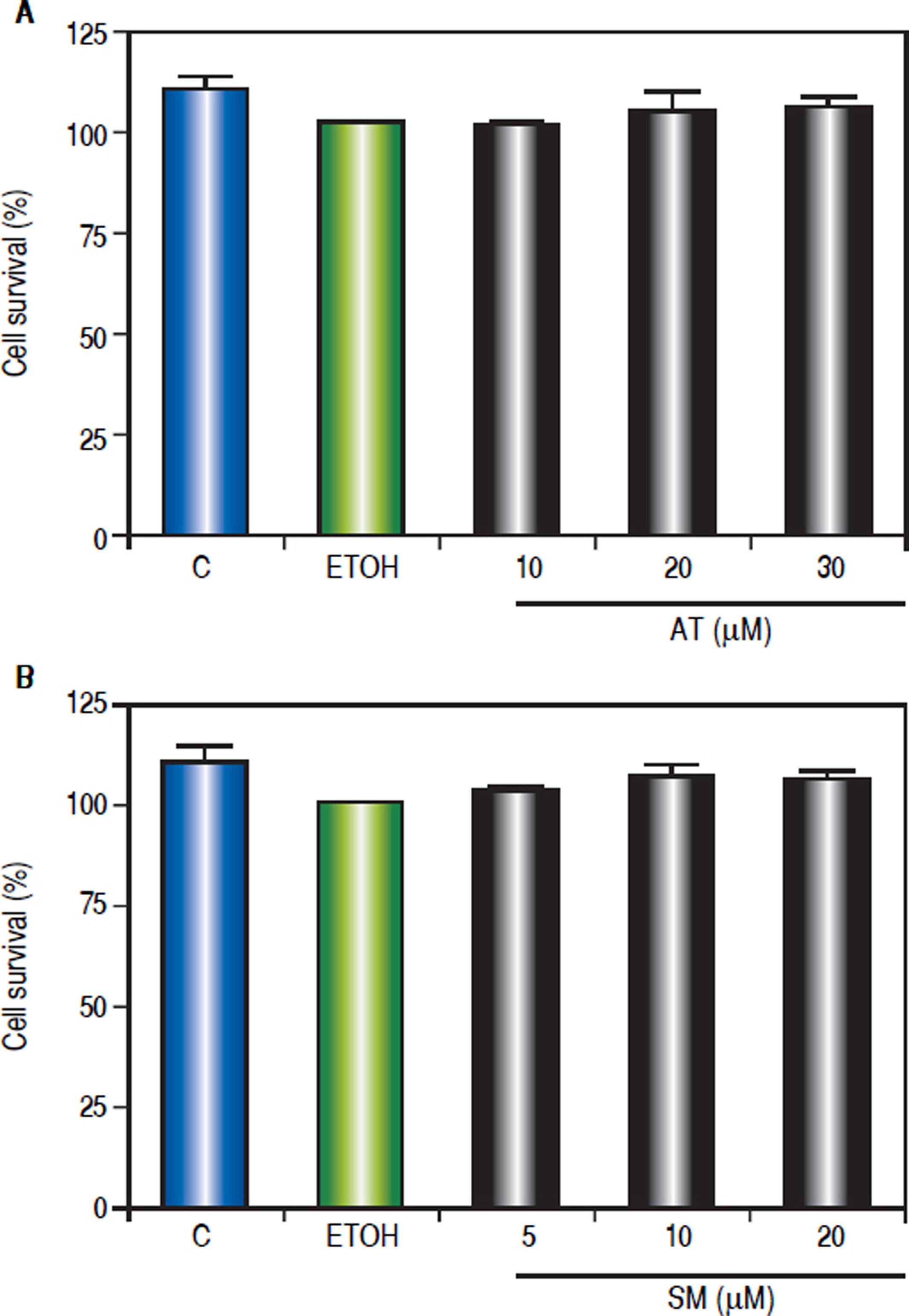
In order to investigate the effect of statins on cell viability, Hep-G2 cells were treated with different doses of AT (10, 20 and 30 μΜ) and SM (5, 10 and 20 μΜ) for three hours. MTT assay shows that statins did not alter Hep-G2 cells viability (Figure 3).
 or (B) SM (5, 10 and 20 μM) for 24 h. Values represent means ± SEM of the three independent studies.")
Effect of statins on Hep-G2 viable cell number. The viable cell number was evaluated using the MTT assay. The absorbance was measured at 570 nm, and the results were expressed as percentage of ETOH-treated cells. A. Cells were treated with AT (10, 20 and 30 μM) or (B) SM (5, 10 and 20 μM) for 24 h. Values represent means ± SEM of the three independent studies.
Hep-G2 is a well-differentiated human hepatocellular carcinoma cell line that preserves numerous liver-specific functions and thus can serve as an in vitro model.
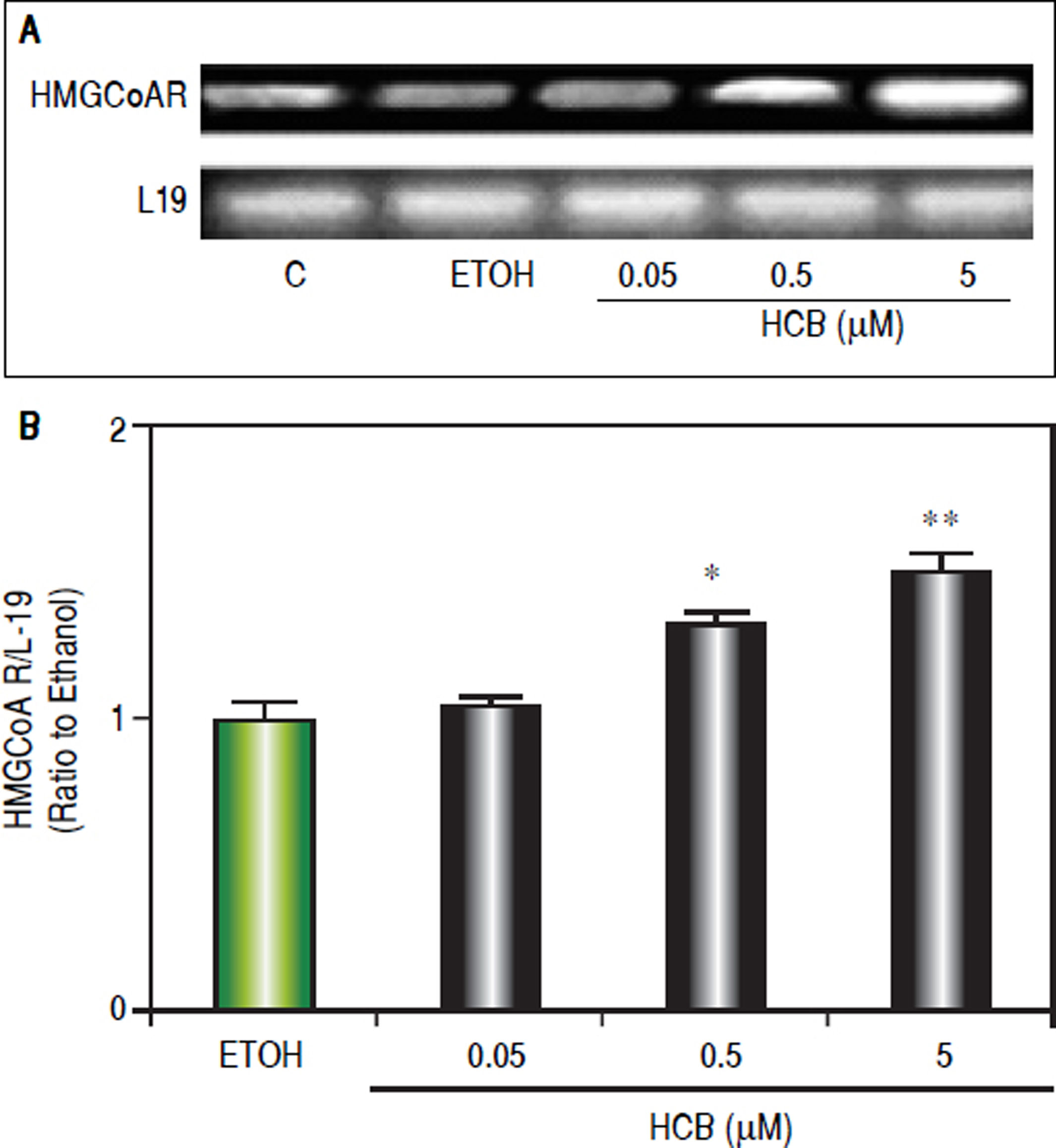
Consistent with our previous results showing that HCB induces cell proliferation in a dose-dependent manner in Hep-G2 cells,26 we evaluated HCB treatment effect (0.05, 0.5 and 5 μΜ) on HMGCoAR mRNA levels. As shown in figure 3, HMGCoAR mRNA levels significantly increased (32% to 54%) in total cell lysates of 0.5 and 5 μΜ HCB-treated cells, respectively, when compared to ETOH (Figure 4).
. A. Representative pattern of HMGCoAR cDNA from ETOH and HCB-treated cells, synthesized from total RNA. L-19 was used as a loading control. B. Quantification of cDNAs, after correction with L-19 cDNA. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05) compared to ETOH group.")
HCB induces HMGCoAR mRNA expression. Hep-G2 cells were treated with HCB (0.05, 0.5 and 5 μM). A. Representative pattern of HMGCoAR cDNA from ETOH and HCB-treated cells, synthesized from total RNA. L-19 was used as a loading control. B. Quantification of cDNAs, after correction with L-19 cDNA. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05) compared to ETOH group.
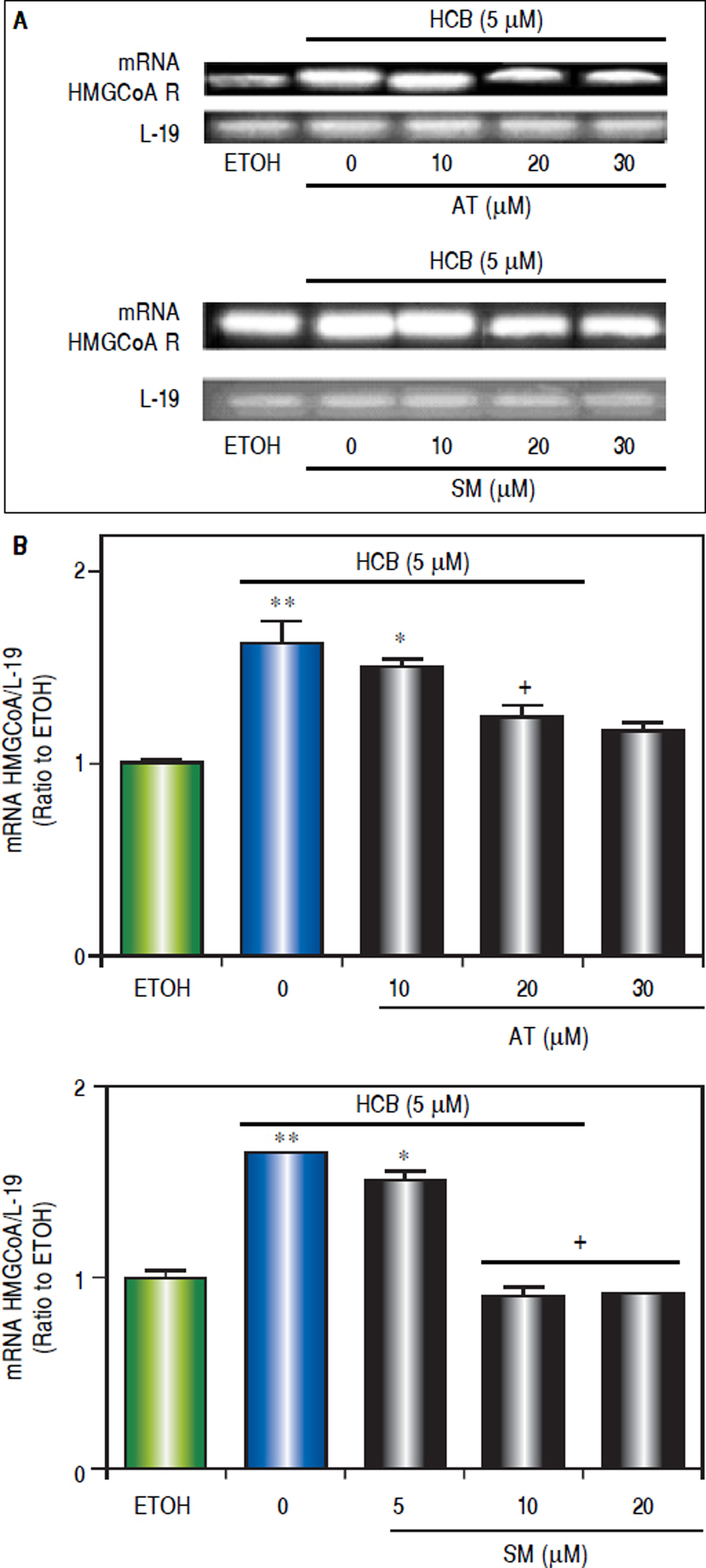
To evaluate the effect of statins on HMGCoAR mRNA expression, Hep-G2 cells were pretreated with increasing doses of AT (10, 20 and 30 μΜ) or SM (5, 10 and 20 μΜ) for 3 h and then treated with HCB (5 μΜ) for 24 h. As shown in figure 5, HMGCoAR mRNA levels significantly increased due to HCB effects. Nevertheless, HMGCoAR mRNA levels significantly decreased by 29% and 38% when pretreated with AT 10 and 20 μM (respectively), reaching baseline values with AT 30 μM, when compared to HCB-treated cells without AT pretreatment. Similarly, pretreatment with SM at 5 and 10 μM decreased HMG-CoAR mRNA levels by 21% and 31% (respectively), reaching baseline values with SM 20 μM, when compared to HCB-treated cells without SM pretreatment (Figure 5).
 or SM (5, 10 and 20 μM) for 3 h and then treated with HCB (5 μM) for 24 h. B. L-19 was used as a loading control. Representative patterns of RT-PCR amplification of HMGCoAR cDNA from ETOH and statinstreated cells, synthesized from total RNA are shown in the upper panel. Quantification of HMGCoAR mRNA, after correction with L-19 is shown in the lower panel. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05) compared to ETOH group.")
Analysis of statins effect on HMGCoA R mRNA expression in HCB-treated Hep-G2 cells. A. Hep-G2 cells were pretreated with AT (10, 20 and 30 μM) or SM (5, 10 and 20 μM) for 3 h and then treated with HCB (5 μM) for 24 h. B. L-19 was used as a loading control. Representative patterns of RT-PCR amplification of HMGCoAR cDNA from ETOH and statinstreated cells, synthesized from total RNA are shown in the upper panel. Quantification of HMGCoAR mRNA, after correction with L-19 is shown in the lower panel. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05) compared to ETOH group.
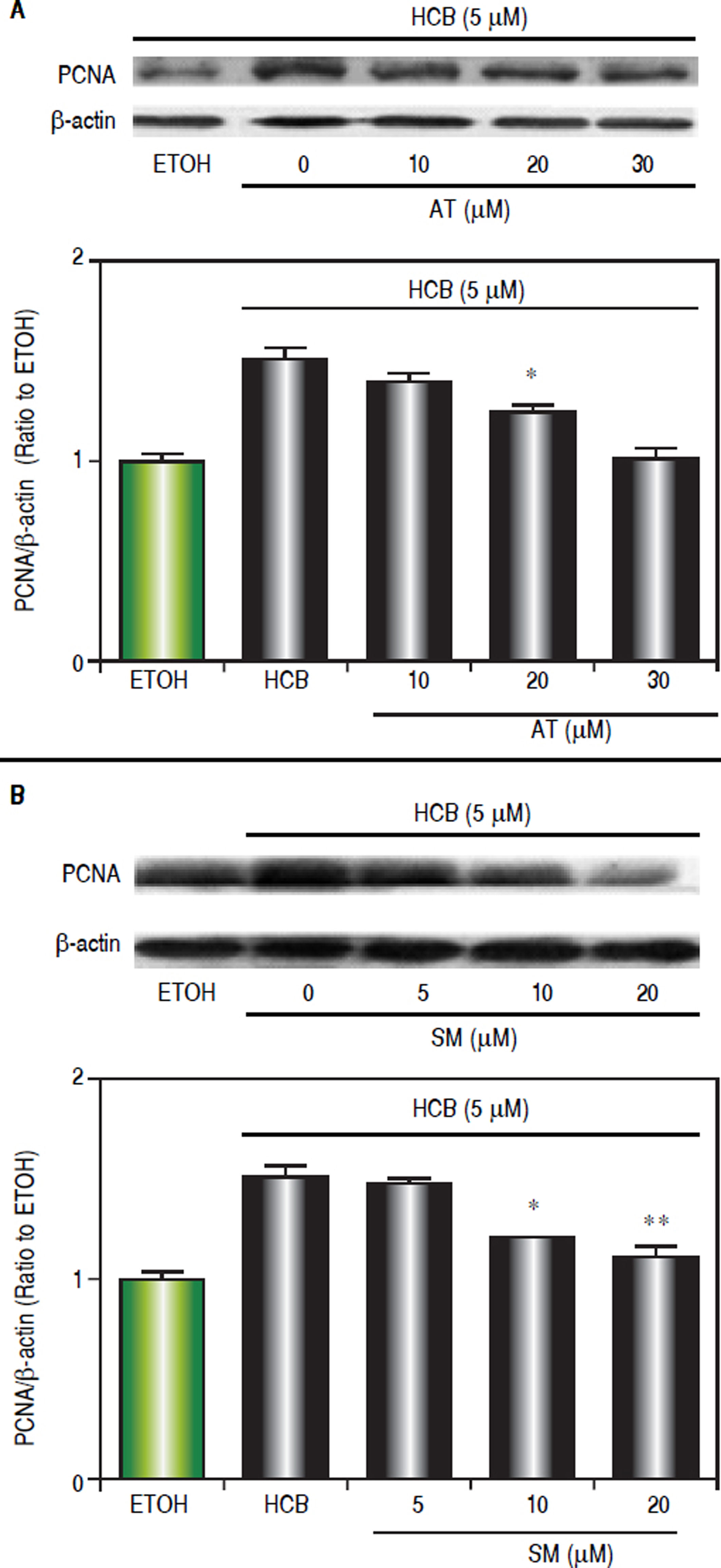
Since we have previously demonstrated that 5 μM HCB induced PCNA-expression in Hep-G2 cells,26 we evaluated the dose-response effects of statins on cell proliferation. Western blots analysis showed that AT pretreatment (10, 20 and 30 μΜ) significantly reduced HCB-induced PCNA levels by 24%, 52% and reaching baseline values at maximum (respectively) when compared to HCB group (Figure 6A). In a similar manner SM pretreatment (5, 10 and 20 μΜ), significantly reduced HCB-induced PCNA levels (30%, 58% until reaching basal values at maximum doses, respectively), when compared to HCB group (Figure 6B). Altogether, the above results demonstrate that statins antagonize HCB-induced cell proliferation in Hep-G2 cells.
 for 3 h. Quantification of PCNA protein levels by densitometric scanning of the immunoblots is shown in the lower panel. B. Western blot analysis of PCNA in total lysate of Hep-G2 cells treated with 5 μΜ HCB, and pretreated with SM (5, 10 and 20 μΜ). Quantification of PCNA protein levels by densitometric scanning of the immunoblots is shown in the lower panel. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05 and **p < 0.01) compared to HCB-treated cells.")
Statins effect on HCB-induced cell proliferation. A. Western blot analysis of PCNA in total lysate of Hep-G2 cells treated with 5 βM HCB, and pretreated with AT (10, 20, and 30 βM) for 3 h. Quantification of PCNA protein levels by densitometric scanning of the immunoblots is shown in the lower panel. B. Western blot analysis of PCNA in total lysate of Hep-G2 cells treated with 5 μΜ HCB, and pretreated with SM (5, 10 and 20 μΜ). Quantification of PCNA protein levels by densitometric scanning of the immunoblots is shown in the lower panel. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05 and **p < 0.01) compared to HCB-treated cells.
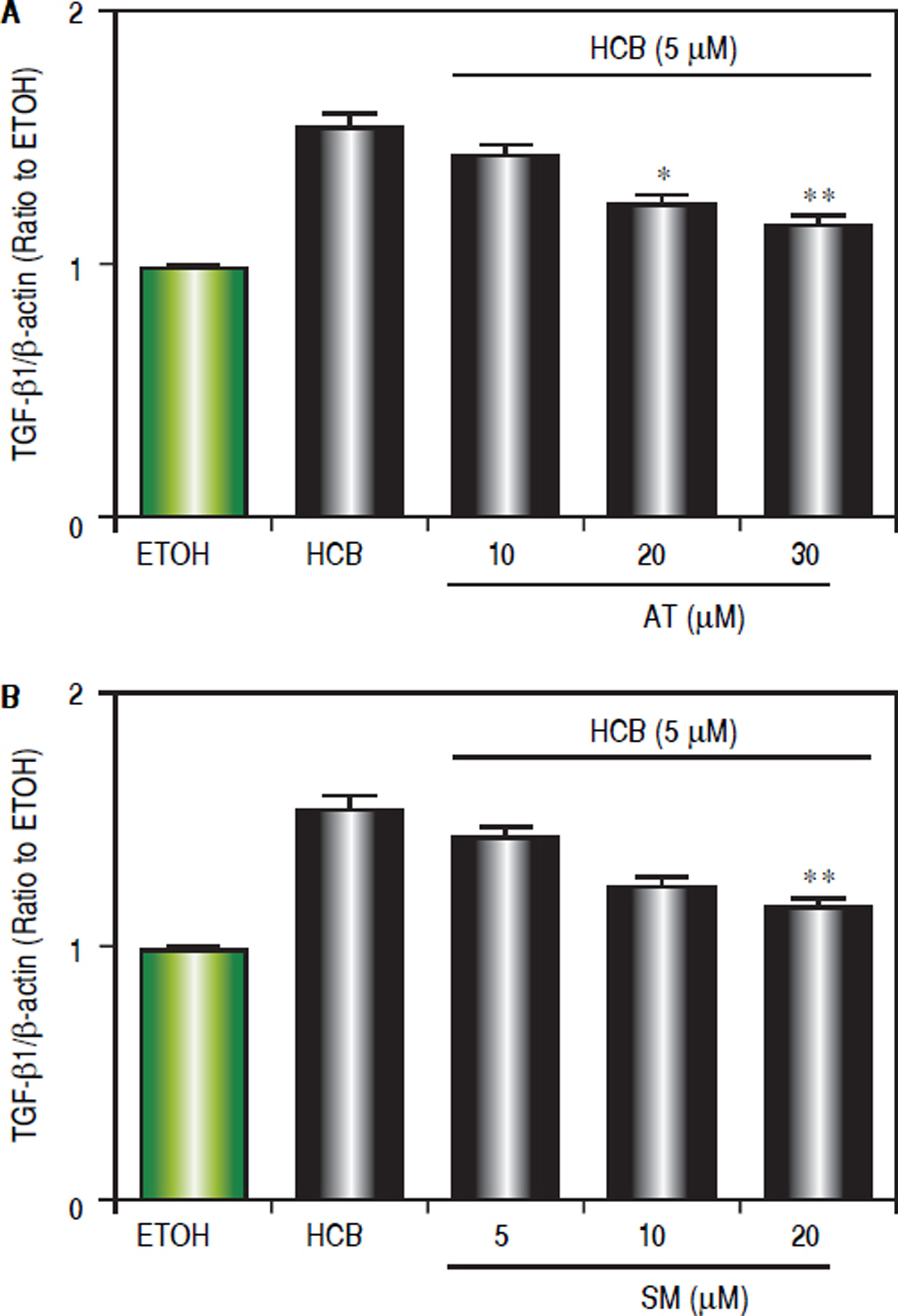
Given that TGF-β1 is one of the most relevant cytokines that intervene in the apoptotic and/or proliferative processes in hepatocytes, we evaluated the statins effect on TGF-β1 levels in HCB-treated Hep-G2 cells. Hep-G2 cells were pretreated with AT (10, 20 and 30 μΜ) or SM (5, 10 and 20 μΜ) or ETOH for 3 h, and then treated with HCB (5 μΜ) for 24 h. Western blot analysis showed a significant reduction in TGF-β1 levels, when Hep-G2 cells were treated with both statins: AT (23%, 29% and 45%), and SM (11%, 24% and 40%) respectively, until reaching baseline values at maximum doses, when compared to HCB group (Figure 7).
 or SM (5, 10 and 20 μM) for 3 h and then treated with HCB (5 μM) for 24 h. Quantification of TGF-β 1 L-19 ratio to ETOH is shown in the lower panel. B. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05 and **p < 0.05) compared to HCB-treated cells.")
Effect of statins on TGF-β 1 protein levels, in Hep-G2 cells treated with HCB. A. Western blot analysis of TGF-β1 protein levels in Hep-G2 cells pretreated with AT (10, 20 and 30 μM) or SM (5, 10 and 20 μM) for 3 h and then treated with HCB (5 μM) for 24 h. Quantification of TGF-β 1 L-19 ratio to ETOH is shown in the lower panel. B. Values are means ± SEM of three independent experiments. Significantly different (*p < 0.05 and **p < 0.05) compared to HCB-treated cells.
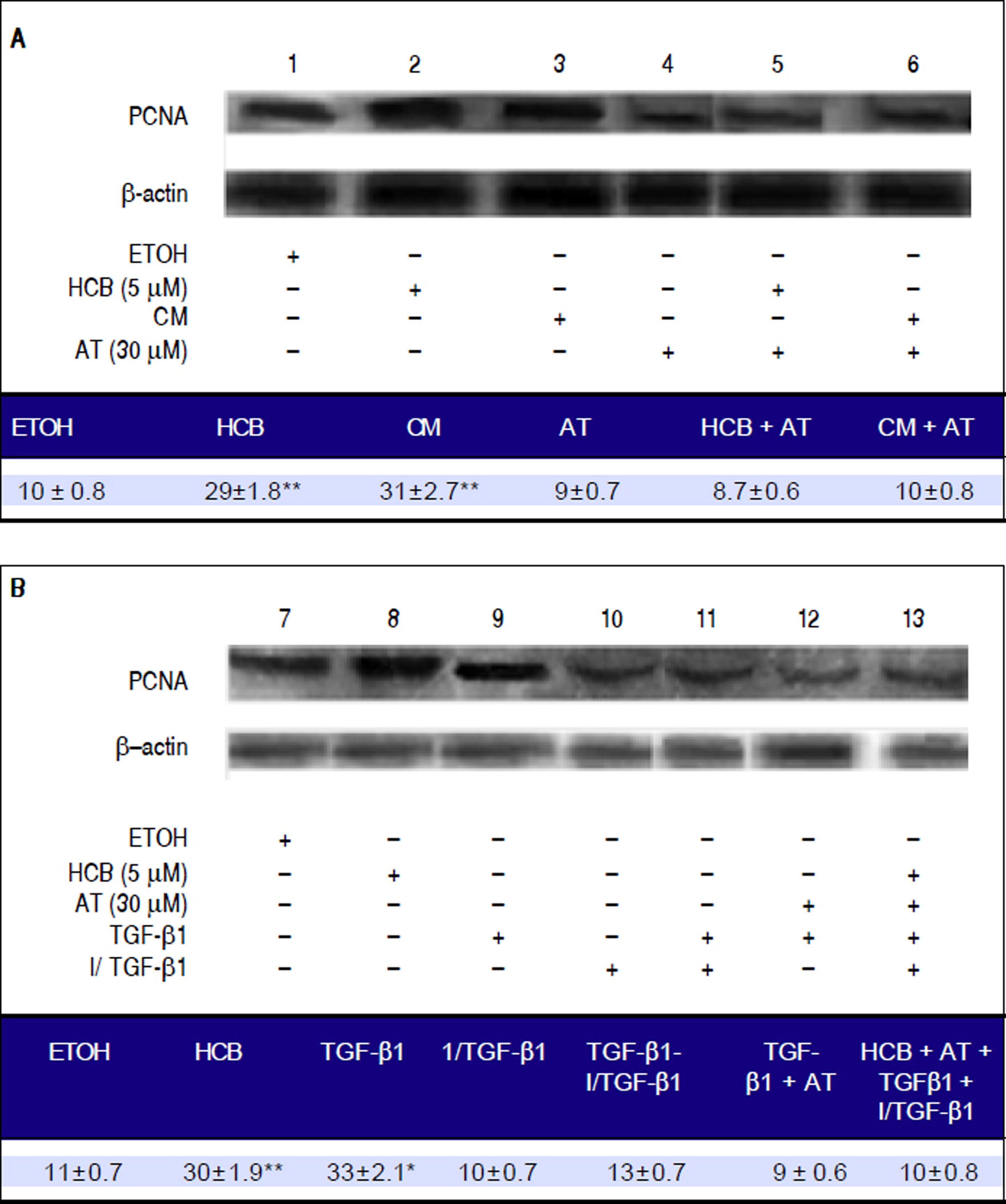
We further investigated whether the statins anti-proliferative effect in HCB-treated Hep-G2 cells was mediated by TGF-β1 When Hep-G2 cells were treated with HCB 5 μΜ (lane 2) or CM (lane 3), a significant increase in PCNA levels (42% and 38%) was observed when compared to ETOH, respectively.
Our results show that AT pretreated cells did not show increases in PCNA levels with HCB or MC treatment (lanes 5 and 6, respectively). On the other hand, Hep-G2 cells treated with TGF-β1 showed a significant increase in PCNA levels (lanes 9) and this effect was blocked when these cells were pretreated with an exogenous TGF-β1 inhibitor (lane 11). Similarly, Hep-G2 cells pre-treated with AT and TGF-β1 showed no changes in PCNA levels (lane 12). Finally, Hep-G2 cells pre-treated with AT and I/TGF-1 and then with an exogenous TGF-β1 and HCB, showed no changes on PCNA levels (lane 13) (Figure 8A and 8B).
, lane 3: Hep-G2 cells treated with conditioned medium (CM), lane 4: Hep-G2 cells pre-treated with AT (30 μM), lane 5: Hep-G2 cells pre-treated with AT + HCB, lane 6: Hep-G2 cells pre-treated with AT + CM, lane 7: Hep-G2 cells treated with ETOH, lane 8: Hep-G2 cells treated with HCB (5 μM), lane 9: Hep-G2 cells treated with TGF-β 1, lane 10: Hep-G2 cells treated with an exogenous TGF-β 1 inhibitor, lane 11: Hep-G2 cells pre-treated with TGF-β1 inhibitor + TGF-β 1, lane 12: Hep-G2 cells pre-treated with TGF-β 1 + AT (30 μM) and lane 13: Hep-G2 cells pre-treated with AT and TGF-β 1 inhibitor and after with HCB and TGF-β 1.")
Role of TGF-β1 in statins-induced antiproliferative effect. Lane1: Hep-G2 cells treated with ETOH, lane 2: Hep-G2 cells treated with HCB (5 μM), lane 3: Hep-G2 cells treated with conditioned medium (CM), lane 4: Hep-G2 cells pre-treated with AT (30 μM), lane 5: Hep-G2 cells pre-treated with AT + HCB, lane 6: Hep-G2 cells pre-treated with AT + CM, lane 7: Hep-G2 cells treated with ETOH, lane 8: Hep-G2 cells treated with HCB (5 μM), lane 9: Hep-G2 cells treated with TGF-β 1, lane 10: Hep-G2 cells treated with an exogenous TGF-β 1 inhibitor, lane 11: Hep-G2 cells pre-treated with TGF-β1 inhibitor + TGF-β 1, lane 12: Hep-G2 cells pre-treated with TGF-β 1 + AT (30 μM) and lane 13: Hep-G2 cells pre-treated with AT and TGF-β 1 inhibitor and after with HCB and TGF-β 1.
Given that TGF-β1 activates c-Src kinase, and c-Src pathway activation has been observed in about 50% of tumors from various organs including the liver,22 we evaluated the effect of statins on c-Src protein levels in HCB-treated cells. Our results showed that HCB significantly increased phosphorylated c-Src protein levels (136%). Pre-treatment with AT (30 μM) and SM (20 μM) decreased this HCB-induced elevation in phosphorylated c-Src levels (41% and 40%, respectively). However, it does not return to baseline values when compared to HCB 5 μM treated cells (Figure 9).
 or SM (20 μM) for 3 h and then HCB (5 μM) for 24 h. β-actin was used as charge control. The results of the bands optical analysis are shown in the bottom panel *p ≤ 0.05, ***p ≤ 0.01 respect HCB.")
Analysis of the expression of c-Src by Western blot. Hep-G2 cells were pretreated with AT (30 μM) or SM (20 μM) for 3 h and then HCB (5 μM) for 24 h. β-actin was used as charge control. The results of the bands optical analysis are shown in the bottom panel *p ≤ 0.05, ***p ≤ 0.01 respect HCB.
Disruptive hormone plays a critical role in the development of pre-neoplastic lesions. T3 is one of the major cell cycle regulators and its levels are regulated by the activity of the enzyme DI. In HCB treated cells, mRNA DI levels were significantly reduced by 45% (p ≤ 0.01). When both statins were added to treated cells, they reversed this effect and mRNA DI levels returned to their baseline value (Figure 10).
Discussion or SM (20 µM) for 3 h and then HCB (5 µM) for 24 h. L-19 was used as charge control. The results of the bands optica analysis are shown in the bottom panel **p ≤ 0.05 respect HCB.")
Dysregulated cell proliferation is the main mechanism of hepatocarcinogenesis, leading to tumor growth and invasion. However, the mechanism of this proliferation is complex and poorly understood. Over the last few years, many molecular targets have been proposed as potential sites for advanced HCC treatment.2,3 Limited clinical and epidemiologic data suggest that statins may improve HCC outcomes, a tumor associated with poor prognosis.4
In this study, we have investigated the cellular effects of statins (atorvastatin and simvastatin) on HCB-induced proliferation in Hep-G2 cells. We found that incubation with atorvastatin or simvastatin inhibits HCB-induced cell proliferation in cultured Hep-G2 cells. We have demonstrated for the first time that, statins induced inhibition of proliferation might at least be partially explained by a decrease in TGF-β1 expression and by a downstream decrease in c-Src phosphorylation.
Other authors observed that simvastatin prevents proliferation and favors cellular apoptosis in a rat osteosarcoma line, UMR-106.27 It is interesting to note that they use doses of 3-10 μM; however, we observed that 20 μM might be an optimal dose for proliferative effect reversion. Atorvastatin inhibits in vitro proliferation at dose of 0.4 nM in hepatic myofibroblasts.28 In this study, although both statins present a complete reversion of the HCB proliferative effect, atorvastatin appears to be more effective at the highest dose (30 μΜ).
In a previous study, we have demonstrated that HCB deregulates cell growth and alters growth factors such as EGF and TGF-β1 expression levels, as well as theirs signaling pathways, both being involved in developmental and tumorigenic processes.26,29 Here, in the in vivo induction/ promotion HCC model and in the in vitro model, we use HCB to induce cell proliferation. This increase in cell proliferation (supported by an increased PCNA levels) was accompanied by increases in HMGCoAR and TGF-β1 mRNA levels.
Previously, we had demonstrated that HCB-induced proliferation in Hep-G2 cell is mediated by an increase in TGF-β1 expression. In this study, we evaluated atorvastatin and simvastatin effect on HCB-induced proliferation. Likewise, we analyzed the potential role of TGF-β1 in the statins mechanism of action in the HCB-induced proliferation model. TGF-β1 involvement in the statins mechanism of action to reduce cell proliferation had been previously described.30,31 However, it is unclear which molecules are involved in the downstream pathway of this cytokine.
One of the potential molecules involved could be c-Src. Its pathway activation has been observed in about 50% of tumors from various organs including the liver,22 c-Src can cooperate with kinases receptors to signal through downstream molecules, such as PI3K/PTEN/Akt, Ras/Raf/ Mek1/2/Erk1/2 and STATs. In many cancer cell lines, tumors signal transducers and transcription activator proteins (STAT) are often deregulated with persistent activation in tyrosine phosphorylation. STAT1 is generally considered a tumour suppressor but there is growing evidence that it can also act as a tumour promoter. Therefore, the JAK/STAT signaling pathway is one of the most promising targets in the cancer therapy. Our hypothesis about the role of cSrc in the inhibition of TGF-β1-induced proliferation is consistent with the observations of Sato, et al., who demonstrated that TGF-β1 can induce c-Src phosphorylation.23
Here, we have demonstrated for the first time, the downstream role of c-Src in Hep-G2 cells proliferation. We have also demonstrated for the first time that TGF-β1/ c-Src pathway may be inhibited by both statins in a dose dependent manner.
The role of thyroid hormones in carcinogenesis has been postulated for many years. However, there is not enough data for a complete understanding of this process. Triiodothyronine (T3) acts through its nuclear receptors, the thyroid hormone receptors (THR). T3 is produced by monodeiodination catalyzed by type 1 iodothyronine deiodinase (DI). This enzyme is present in almost all tissues, but the highest concentrations were found in the thyroid, the liver, and the kidneys.32 Both its enzymatic activity and its gene expression have been examined in some neoplastic tissues. Significant decreases in gene expression and enzyme activity were found in several of them.33 HCB is a hormone disruptor and induces hypothyroxinemia, and it is well known that T3 has a negative regulatory effect on the cell cycle.29 A possible explanation could be that thyroid hormones deregulation may play an important role in HCC development as well as in the statins mechanism key on cell proliferation inhibition.
In our previous study, we demonstrated that HCB increases cell proliferation, alters mRNA DIII and TH concentration in induction/promotion rat liver model.29 In this study, our results showed that HCB increased cell proliferation and decreased mRNA DI levels in Hep-G2 cells. It is possible that DI decreased expression is partly responsible for the decrease in tissue T3 and this event contributes to the deregulation of the cell cycle. More experiments are needed to explore this finding.
We showed here that the increase in cell proliferation as well as the decrease in mRNA DI levels are mediated by TGF-β1 We also demonstrated that statins inhibit cell proliferation by decreasing TGF-β1 levels.
It is known that there is a regulation between TGF-β1 and DI enzyme. It is possible that statins mediated TGF-β1 reduction counteracts the effects on DI and T3 levels, resulting in normal T3 tissue levels.
In preliminary experiments (data not shown) performed in our laboratory, we observed that exogenously administered thyroid hormones reduce HCB-induced cell proliferation in the Hep-G2 cell line. Won Park, et al. showed that THR could down regulate the activity of cSrc in the presence of T3 via phosphorylation, suggesting that low T3 levels generate a c-Src related pro-proliferative process.34 More experiments are needed to validate these results in our experimental model.
Summarizing, our study showed for the first time that atorvastatin and simvastatin can revert HCB-induced proliferative effects on Hep-G2 cells. In addition, this effect is mediated by the inhibition of TGF-β1 and the downstream c-Src phosphorylation pathway. The TH homeostasis deregulation could be potentially involved. TGF-β1 and c-Src may be the statins molecular targets in early events of hepatocarcinogenesis.
Abbreviations- •
AT: Atorvastatin.
- •
CM: conditioned medium.
- •
c-Src: cellular Src kinase, Proto-oncogene tyrosineprotein kinase Src.
- •
DEN: Diethylnitrosamine.
- •
DI: deiodinase I.
- •
HBV: hepatitis B virus.
- •
HCB: hexachlorobenzene.
- •
HCC: Hepatocellular carcinoma.
- •
HCV: hepatitis C virus.
- •
HMGCoAR: 3 -hydroxy-3-methylglutaryl coenzyme-A reductase.
- •
HSCs: hepatic stellate cells.
- •
HVPG: hepatic venous pressure gradient.
- •
i.p.: intra peritoneal.
- •
PCNA: Proliferating cell nuclear antigen.
- •
pc-Src: phosphorylated c-SRC.
- •
SM: Simvastatin.
- •
STAT: signal transducer and activator of transcription.
- •
T3: triiodothyronine.
- •
TGF-β1: transforming growth factor-β1.
- •
TH: thyroid hormones.
- •
THR: thyroid hormone receptor.
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Financial SupportThis work was supported by grant SAF2012-32491 from Plan Nacional (MINECO) and grant S2010/BDM-2423 (MOIR) from Comunidad de Madrid, Spain, and by a grant UBACYT 20020120200258 /EXP-UBA N° 17064/ 2012 from Universidad de Buenos Aires, Ciudad Autónoma de Buenos Aires, Argentina.



