






IgG4-related disease is a recently-described fibro-inflammatory condition with characteristic histopathological findings in the organs involved. The most commonly affected organs are pancreas, lymph nodes, and retroperitoneum. Liver disease usually involves bile structures and therefore IgG4-related disease is considered a cause of secondary sclerosing cholangitis. One out of three patients with IgG4 sclerosing cholangitis also presents autoimmune pancreatitis, although it can be associated with manifestations in other organs. One of the main features of IgG4-related disease is its good prognosis due to the great response to glucocorticoid therapy. However, relapse of the disease is not uncommon, especially when steroid therapy is decreased or stopped. Rituximab seems to be an effective treatment to achieve remission of the disease. We report the case of a 74 year-old man diagnosed with IgG4-related disease based on increase of serum IgG4 levels, imaging and histopathological findings, with systemic involvement including sclerosing cholangitis. Despite the absence of liver fibrosis at onset, the early use of glucocorticoids and rituximab therapy, the patient presented clinical and analytical deterioration, leading to secondary biliary cirrhosis. In conclusion, this clinical case highlights the importance of prompt diagnosis and therapeutics for sclerosing cholangitis secondary to IgG4-related disease in order to avoid progression of the disease and development of liver cirrhosis, as well as the refractory, aggressive nature of the disease in some cases as this one.
Immunoglobulin G4-related disease (IgG4-RD) has become within recent years a well-established fibro-inflammatory disorder.1 The most commonly affected organs are pancreas, lymph nodes, lacrimal and salivary glands as well as the retroperitoneum.2 Liver disease usually involves bile duct structures and, for this reason, IgG4-RD is considered an important cause of secondary sclerosing cholangitis (SSC). IgG4-SC occurs in a third of patients with type 1 autoimmune pancreatitis (AIP),3 but other organ involvement can be possible. IgG4-RD is considered an entity with a relatively good prognosis due to its high rate of response to steroid therapy.4-6 However, since relapse after stopping steroid treatment or dose reduction is frequent,2 other immunosuppressive agents have been explored.7 In this regard, rituximab has shown its role in a clinical trial where sustained disease response was achieved in 22 out of 30 patients, including patients with biliary involvement.8 Herein we describe a case of systemic IgG4-RD (biliary tract, lymph nodes, bone) that progressed to secondary biliary cirrhosis despite its early diagnosis and treatment with steroids and rituximab, leading to the question if more aggressive or different management is needed in patients with IgG4-RD with biliary strictures in order to avoid fibrosis progression.
Case PresentationA 74 year-old Caucasian man was admitted to emergency department due to daily fever, weight loss, and jaundice over the previous 2 months. He had been reported 10 kg weight loss during the previous two months, as well as hyporexia, fatigue, daily fever up to 38.5°C and right upper quadrant abdominal pain. In addition, he also presented jaundice and white stools the previous week to admission to hospital.
His medical history included arterial hypertension, asthma and pulmonary emphysema, hyperuricemia with occasional gout attacks, benign prostatic hyperplasia and palmoplantar psoriasis. He gave up smoking 15 years ago and he had had a history of 30 pack-years of tobacco use. He referred daily intake of 30 g of alcohol, without history of drug abuse. The patient worked as chemical engineer until his retirement. He presented occupational exposure to silica dust. Regarding family history, his brother died from lymphoma at the age of 71 years.
On physical examination, he had a fever (38°C), jaundiced skin, bilateral axillary lymph nodes, regular heart rate and rhythm without murmurs, but crackles in both low lung fields. Abdomen was soft, nondistended with tender hepatomegaly. Vital signs were stable.
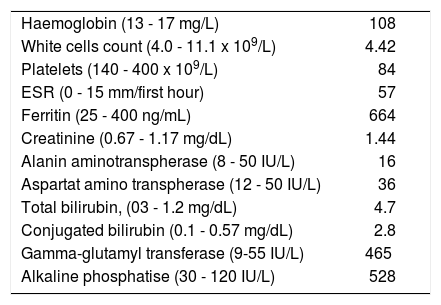
Blood test revealed increased cholestasis enzymes and acute phase reactants, and mild anemia (Table 1). Quantification of immunoglobulins showed increased levels of IgG (4,226 mg/dL). Viral serology (HBsAg, anti-HCV, hepatitis E virus IgM, HIV, EBV IgM and CMV Ig M) were all negative. Anti-mitochondrial antibodies, anti-nuclear antibodies, anti-liver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies (ANCA) were negative.
Laboratory data at diagnosis.
| Haemoglobin (13 - 17 mg/L) | 108 |
| White cells count (4.0 - 11.1 x 109/L) | 4.42 |
| Platelets (140 - 400 x 109/L) | 84 |
| ESR (0 - 15 mm/first hour) | 57 |
| Ferritin (25 - 400 ng/mL) | 664 |
| Creatinine (0.67 - 1.17 mg/dL) | 1.44 |
| Alanin aminotranspherase (8 - 50 IU/L) | 16 |
| Aspartat amino transpherase (12 - 50 IU/L) | 36 |
| Total bilirubin, (03 - 1.2 mg/dL) | 4.7 |
| Conjugated bilirubin (0.1 - 0.57 mg/dL) | 2.8 |
| Gamma-glutamyl transferase (9-55 IU/L) | 465 |
| Alkaline phosphatise (30 - 120 IU/L) | 528 |
A magnetic resonance cholangiopancreatography (MRCP) was performed demonstrating a hepatosplenomegaly, irregularities in intra-and extrahepatic bile ducts and multiple enlarged mesenteric and hilar lymph nodes. No pancreatic enlargement or narrowing of the main pancreatic duct was observed. At the positron emission tomography abnormal uptake was presented at liver, spleen, supra and infradiaphragmatic lymph nodes (Figure 1).

In order to achieve a definitive diagnosis, biopsies of liver and an axilar lymph node were carried out. They showed intense lymphoplasmacytic inflammatory infiltrate with an increased number of plasma cells, as well as eosinophils and an increase IgG4+cell/IgG+ cell ratio of 74% and 65% in liver and lymph node, respectively (Figure 2A and Figure 2B). No granulomas were observed. Only mild fibrosis was observed in liver biopsy (METAVIR score 0-1). Peripheral blood immunophenotype showed that 9.7% of the total B-cells were plasmablasts (normal range 0.1-1.5%) and blood IgG4 level was increased (1,900 mg/dL, normal range 5-156 mg/dL). Liver and lymph node biopsies were highly suggestive of IgG4-RD (IgG4 + cell/IgG+ cell ratio 74% and 65%, respectively). Moreover, the biliary tract imaging and elevated serum IgG4 concentration supported the definitive IgG4-RD diagnosis according to the 2012 international consensus criteria.9
 and axilar lymph node (B) biopsy showed an intense limphoplasmocitary inflammatory infiltrate with an increased number of plasmatic cells, eosinophils, and an increased IgG4 + cell/IgG + cell ratio (74 and 65%, respectively).")
Six days after admission, steroids were initiated at dose of 1 mg/kg/day. The patient presented rapid improvement of general condition and jaundiced skin, with remission of fever within the next 24 h. However, 6 weeks after progressively tapering of the steroids dose, he once again presented fever and jaundice. For this reason, rituximab was given in two doses of 1,000 mg, administered 15 days apart, achieving a complete B-cell depletion. Low dose of steroids (10 mg/day) were maintained. Nevertheless, approximately one month after rituximab therapy, the patient exhibited clinical deterioration with reappearance of fatigue, fever and jaundice (serum bilirubin 4.5 mg/dL), and for this reason, the steroid dose was increased and azathio-prine at a dose of 2 mg/kg/day was added. Despite steroids pulse therapy (1 mg/kg/day and afterward dose of 30 mg/ day), bilirubin levels continued progressively increasing up to 17 mg/dL. A new MRCP showed worsening of the thickening of both intra and extrahepatic biliary tract (Figure 3). In conjunction with the Radiology and Endoscopy Departments the possibility of endoscopic retrograde cholangio-pancreatography was ruled out given the large and diffuse extension of structures, especially in the intrahepatic ducts. A new liver biopsy 6 months after the diagnosis showed improvement in IgG4 + cell/IgG + cell ratio but appearance of collagen bridges, suggesting an emergent secondary biliary cirrhosis. Moreover, a gastros copy showed signs of portal gastropathy. The patient’s clinical situation deteriorated progressively, and finally, the patient passed away 8 month after IgG4-RD diagnosis as consequences of multiorgan failure due to septic shock by Serratia marcescens.
Discussion and control (B) MRCP showed marked worsening of the diffuse bile duct thickening, i.e. at the common bile duct (white arrows).")
IgG4-RD is a fibro-inflamatory condition comprising a group of entities that were considered non-related in old time, but in which specific pathologic findings are constantly present. These are the presence of lymphoplasmacytic infiltrates, storiform fibrosis and obliterative phlebitis in the context of significant IgG4 + plasma cell infiltrates. A common clinical feature is a tendency to present clinically with a pseudo-tumor in the involved organs. Up to 45-50% of patients, depending on the series,2,10 have a systemic involvement with more than 1 organ affected, a fact that may be helpful for the suspicion of this disease. Moreover, it is frequently associated with type I AIP. It can also involve the biliary tract, so in recent years, IgG4-RD has been added to the list of diseases that can lead to SCC.
An important point is to differentiate between primary and SCC. Primary sclerosing cholangitis (PSC) is a chronic, cholestatic liver disease that leads to irregular obliteration of both intra and extrahepatic bile ducts.11 Up to 80% of PSC patients have concomitant inflammatory bowel disease that in the majority of cases is diagnosed with ulcerative colitis.12 Most patients are relatively young at onset (30-40 years of age).13 A diagnosis of PSC is made in patients with elevated serum markers of cholestasis not otherwise explained and typical cholangiographic findings. Both the American and European guidelines11,14 recommend MRCP as the first option in cases of suspected PSC, and endoscopic cholangiography for non-diagnostic cases. Liver histological findings may support a diagnosis of PSC, especially in those cases where small duct are affected presenting with typical periductal concentric “onion skin” fibrosis.12 The typical MRCP features include diffuse, multifocal short segmental strictures and mild dilatation in the intrahepatic and extrahepatic bile ducts alternating with normal ducts, which sometimes produce a “beaded” appearance. As the fibrosis progresses and the strictures worsen, the peripheral bile ducts are obliterated and become poorly visualized on MRCP, showing a “pruned tree” appearance. The autoantibodies most frequently reported are perinuclear ANCA (pANCA) (26-94%). A variety of other autoimmune diseases (e.g. type 1 diabetes, thyroid disease, psoriasis, coeliac disease, etc.) have also been reported at an increased frequency in patients with PSC.13
Our patient presented irregularities at intra and extrahepatic bile ducts. However, he was relatively elderly at onset of the symptoms and he did not suffer from inflammatory bowel disease. Moreover, ANCA were negative, and the liver biopsy ruled out small duct PSC. Moreover, the marked increased levels of IgG4 plus the imaging and histolopathological finding led to the definitive diagnosis of IgG4-SC.
An important issue for the diagnosis of IgG4RD is that 50-60% of patients have raised levels of IgG4 in serum. Actually, IgG4 serum concentrations can be useful for differentiation between PCS and IgG4-SC.15 The clinical picture is dominated by obstructive jaundice, reflecting marked concentric stenosis of the large bile duct. Elevated serum concentrations of IgG4 are found in up to 60% of patients and a IgG4 + cell/IgG + cell ratio in tissues > 40% had a sensitivity and specificity of 94.4% and 85.7%, respectively. Due to the difficulties with IgG4-RD diagnosis, in 2012 international criteria including clinical, histopathological and imaging findings were established.
The majority of patients with IgG4-RD respond to steroids, especially in early stages of the disease. However some of them can relapse and given the potential complications that can appear in the context of this disease, the use of immunosuppressive or even biological agents, e.g. rituximab, should be considered.16,17 Although in the experience from Mayo Clinic, the relapse-free survival in AIP patients treated with steroids alone vs. steroids plus an immunomodulator (azathioprine, 6-mercaptopurine or mycophenolate mofetil) was similar, ten out of 12 subjects treated with rituximab achieved complete remission.7 Moreover, the majority of patients with biliary strictures presented complete remission and only one case persisted with stable disease.7 More recently, an open label trial including 30 IgG4-RD patients treated with rituximab showed 97% of disease response within first 6 months after treatment.8 Ten (33%) subjects presented biliary involvement but none relapsed after rituximab therapy.
In our case, after definitive diagnosis of IgG4-RD, based on an increase of serum IgG4 levels, imaging and histopathological findings, steroids therapy was initiated and afterward rituximab was administrated, given the relapse of symptoms and analytical worsening after the steroids dose was tapered. Reappearance ofjaundice despite B-cell depletion led to the performance of a new MRCP that showed diffuse thickening of both intra and extrahepatic biliary tract. Moreover, a liver biopsy confirmed the presence of signs of secondary biliary cirrhosis despite the decrease in IgG4 + cell/IgG + cell ratio, and the improvement in IgG4-RD index activity. Data from patients with AIP pointed out that obstructive jaundice at onset was an independent risk factor for unfavourable events (HR 3.09).18 On the other hand, early steroids treatment acted as a protective factor (HR 0.33).18 Although our patient presented unfavourable factors for relapse, in previous series rituximab therapy allowed the removal of biliary stents in patients with IgG4-SC.3,7 Management of patients with IgG4-SC with diffuse biliary tract involvement may include early rituximab therapy in order to avoid fibrosis development or even placement of prophylactic stents so as to improve outcome since the disease may otherwise rapidly progress with serious complications.
In conclusion, herein we present an atypical case of Ig4-related disease, considering the systemic manifestations and the poor clinical and analytical course despite early therapy with steroids and rituximab. The facts which provided clues for the diagnosis were the radiological findings in the biliary tree and the presence of lymph nodes. Both liver and lymph node biopsies supplied the definitive diagnosis. Despite steroids pulse therapy and rituximab, the patient presented poor outcome with development of liver injury. This clinical case highlights the importance of prompt diagnosis and therapeutics for sclerosing cholangitis secondary to IgG4-related disease in order to try to prevent progression of the disease and development of secondary biliary cirrhosis.
Abbreviations- •
IgG4-RD: immunoglobulin G4-related disease.
- •
SSC: secondary sclerosing cholangitis.
- •
AIP: autoimmune pancreatitis.
- •
ANCA: antineutrophil cytoplasmic antibodies.
- •
MRCP: magnetic resonance cholangiopancreatography.
- •
PSC: Primary sclerosing cholangitis.
- •
IgG4-SC: IgG4-sclerosing cholangitis.
The authors declares that there is no conflict of interest regarding the publication of this article.
FundingThe authors declare that they have received no funding for this manuscript.
AcknowledgmentsEnglish writing support was provided by Fidelma Greaves.



