




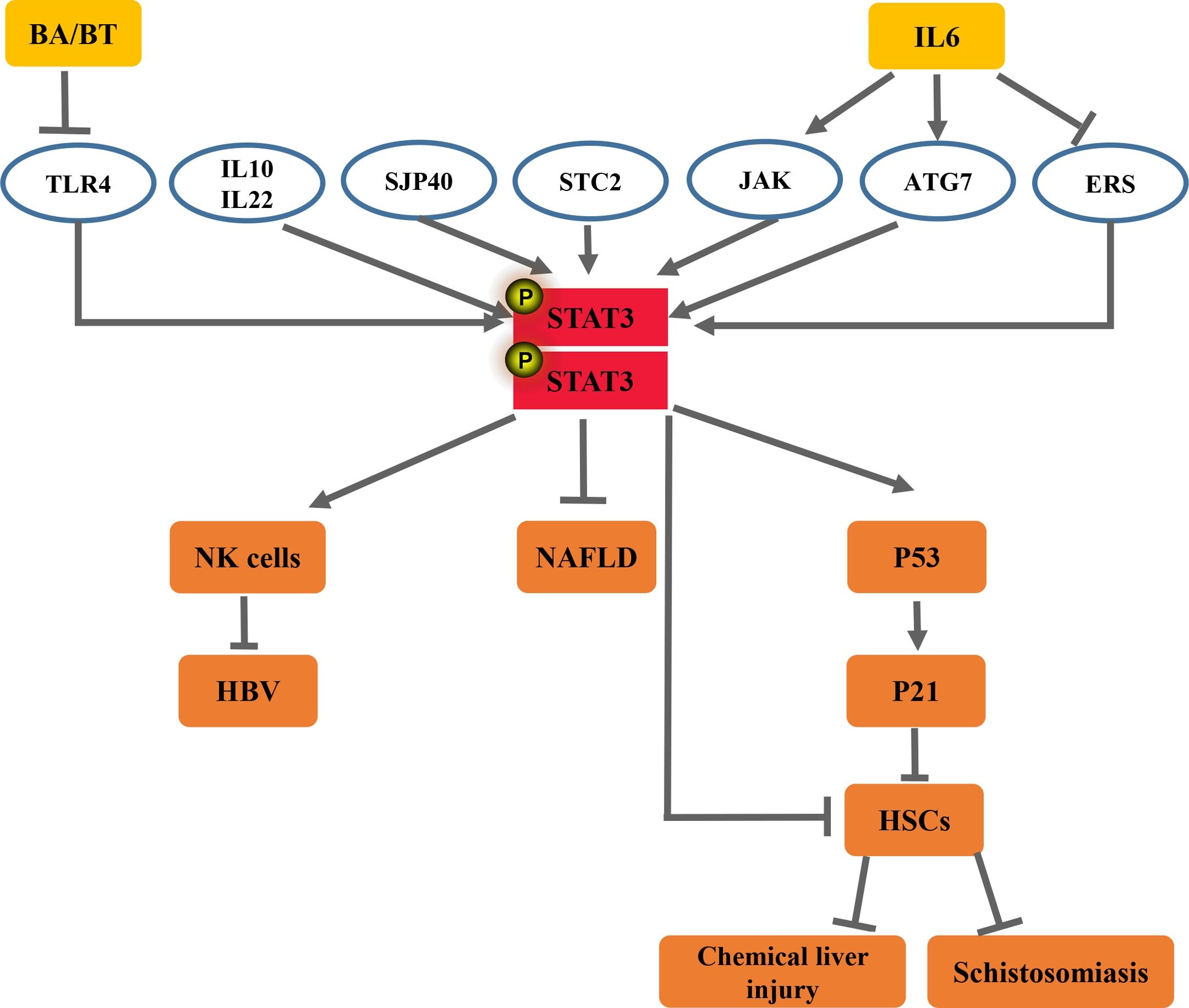
Janus protein tyrosine kinase (JAK) has the ability to activate signal transducer and activator of transcription (STAT). STAT3 is a valued member of the JAK/STAT signaling pathway. In recent years, several studies have documented that STAT3 is closely related to the occurrence and development of liver fibrosis caused by various factors. Activation of STAT3 can play anti- or pro-inflammatory roles in the pathogenesis of liver fibrosis. This article reviewed the recent studies on STAT3 in the development of various liver fibrosis to find a more effective method to relieve and cure liver diseases, such as hepatitis B virus (HBV), non-alcoholic fatty liver disease (NAFLD), schistosomiasis, and chemical liver injury.
Liver fibrosis results in the excess and reversible accumulation of fibrillar extracellular matrix (ECM) components in the liver, which is a major determinant of reiterated liver tissue damage because of various predisposing factors, such as hepatitis B virus (HBV) and hepatitis C virus, alcohol consumption, non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), parasitic infection (Schistosoma), and drug/chemical toxic liver injury [1]. Activation of hepatic stellate cells (HSCs) is regarded as the pivotal link to liver fibrosis. When the liver is injured, the quiescent HSCs are activated and then transformed into myofibroblasts, which secrete a large number of ECM components, leading to excessive deposition of ECM. The accumulation of ECM is quite frequent leading to an abnormal wound healing response [1,2,3]. In this process, many molecules were identified in the regulation of the fibrous scar formation.
STAT3 plays a pivotal role in the pathogenesis of liver diseases [4]. STAT3 is reported to be a cytoplasmic signal transcription factor that belongs to the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway and plays a crucial role in the process of mediating liver injury. Many cytokines and growth factors through the JAK-STAT pathway, an important regulatory pathway, play a vital role in mammals [4]. In general, receptors binding and subsequent receptor induced by cytokines dimerization can contribute to receptor-associated JAK dimerization. Activation of the JAK triggers phosphorylation of specific tyrosine residue receptors. Phosphorylated tyrosine sites subsequently form specific “docking sites” with surrounding amino acid sequences, STAT proteins are recruited to this “docking site”. The STAT protein has been immediately phosphorylated, and then the activated STAT protein translocates to the nucleus as a dimer to bind to the target gene and modulates gene transcriptional control [4,5,6]. STAT3 can be activated by over 40 peptide hormones, of which the best examined are members of the interleukin (IL)-6 family of cytokines [7]. The STAT3 pathway appears to play a controversial role in liver fibrogenesis due to the recorded hepatoprotective and proliferative functions of STAT3 [8,9].
STAT3 has a positive role in regulating many aspects of cell survival and proliferation. However, when it is persistently activated, the deleterious effects lead to various pathological manifestations [7,10]. Thus, these findings suggest that persistent activation of STAT3 plays a crucial role in liver fibrogenesis. Activation of STAT3 has been detected in many fibrotic tissues [7,11,12]. However, the possible molecular mechanisms that elucidate the role of STAT3 in the initiation and progression of liver fibrosis are still not fully understood.
In this review, we highlight the various vital functions of STAT3 in viral hepatitis, alcohol consumption, NASH, NAFLD, drug/chemical toxic liver injury, and parasitic infection.
2The role of STAT3 in HBVHBV has a circular, partially double-stranded DNA that belongs to the hepatotropic virus, which can cause severe liver diseases, including acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC). Globally, approximately 250 million people are suffering from chronic HBV infection, contributing to nearly one million deaths annually, which are a global public health problem [13]. HBV is a member of the hepadnavirus family with a compact DNA genome replicating by the viral reverse transcriptase from an RNA intermediate [14,15]. Under the electron microscope, HBV can exhibit three particle structures: large spherical particles with a diameter of approximately 42 nm, small spherical particles with a diameter of approximately 22 nm, and tubular particles. The virus genome contains four open reading frames encoding seven proteins: three surface proteins (LHBs, MHBs, and SHBs), hepatitis B core antigen, hepatitis B e antigen, the viral polymerase, and the X protein [14,16].
The post-necrotic or bridging fibrosis is the prevalent pattern of fibrosis evolution in HBV-induced liver fibrosis, and increased deposition of ECM components are key features. The prevailing pro-fibrogenic mechanism in HBV-induced liver fibrosis is represented by the chronic response of wound healing, with liver parenchymal injury, hepatocyte death, and oxidative stress also related to fibrosis. HSCs are transformed into myofibroblasts after activation and then secrete a large amount of ECM, which is the most important pathological feature of liver fibrosis [17].
Cellular factors or cytokines participate in transcriptional activities of HBV promoters, including hepatocyte nuclear factor 4, peroxisome proliferator-activated receptor, retinoid X receptor, and STAT3 [14,18].
STAT3: an anti- and pro-inflammatory signal in HBV infection. In HBV infection, STAT3 activation promotes or reduces liver fibrosis depending on the progression of liver injury.
2.1Anti-inflammatory signalContemporary studies suggest that STAT3 is an important connective regulator between HBV persistence and natural killer (NK) cell dysfunction. NK cells are essential components of the innate immune system, which can inhibit the progression of liver fibrosis. Infection-induced STAT3 downregulation resulted in NK dysfunction and diminished its response to IL-21, whereas IL-21 promotes the ability of NK cells to kill abnormal cells. In addition, STAT3 activates NK cell functions via directly targeting the NK cell activating receptor, NKp46. whereas, exogenous STAT3 enhancement reversed the NK dysfunction induced by anti-chronic HBV (CHB) infection, diminishing the liver fibrosis. These findings reveal that a novel mechanism of STAT3 down-regulation results in NK dysfunction during CHB infection and aggravates liver fibrosis induced by HBV [19].
A growing body of evidence suggests that several complex formulas and crude materials used in Chinese medicines have measurable activity against viral hepatitis, including Xiaochaihu decoction (XCHD) [20]. The study demonstrated that culture in serum containing modified XCHD stimulated hepatocyte proliferation and favored the inhibition of Hepatitis B surface antigen (HBsAg) by contributing to the expression of STAT3 [20]. These findings have documented that the protective effect on chronic liver fibrosis might be because of the activation of STAT3. STAT3 has a regulatory role in protection of the liver, which merits further investigation.
2.2Pro-inflammatory signalAccumulated research data from the last decade suggest that STAT3 plays key roles in accelerating liver injury, fibrosis, and inflammation. The homeobox domain of homeobox protein A10 (HoxA10) is structurally related to the helix-turn-helix motif of prokaryotic DNA-binding proteins, which regulates gene expression during differentiation and cancer progression [14,21]. The data suggest that HoxA10 plays a host negative regulator role to attenuate HBV replication through accelerating the dephosphorylation of the p38 mitogen-activated protein kinase (MAPK)/STAT3 pathway by two approaches. In addition, HoxA10 directly binds to the HBV liver-specific enhancer element I (EnhI)/X promoter and competes with STAT3 for binding to the promoter and thereby represses HBV transcription [14]. Thus, the blockading of hepatic STAT3 activation via HoxA10 prevented HBV-induced liver injury, whereas IL-6+ radiotherapy induced HBV reactivation via interaction of p-STAT3/HNF-3 protein complex and EnhI in HBV transgenic mice [22]. In addition, recent studies have revealed that the expression of miRNA-340 (miR-340) is downregulated in several types of cancer, indicating that miR-340 has an anti-cancer effect and is related to cell differentiation, proliferation, apoptosis, and tumor cell migration and invasion [23,24]. Xiong et al discovered that HBV increases the migration of liver cancer cells, which is likely mediated by downregulating miR-340-5p expression to induce STAT3 overexpression [23]. Therefore, the activation of STAT3 can accelerate HBV-induced liver fibrosis and liver cancer.
The pro-inflammatory roles of hepatic STAT3 in liver fibrosis also have been extensively characterized in another way. Myeloid-derived suppressor cells (MDSCs) are negative regulators of immune responses by weakening T cell proliferation, antibody and interferon-γ production, and CTL induction [25,26]. HBsAg could impair T cell activation by polarizing monocytes toward MDSCs in an extracellular modulated protein kinases/IL-6/STAT3 signaling-dependent manner, which accelerates the liver injury [25]. Oxidative stress may be one of the mechanisms in charge of the activation of protein kinase B and STAT3 in HBV/HCV-expressing HCC cells [27].
In general, STAT3 activation in HBV acts as a pro-inflammatory signal to promote liver fibrosis and even liver cancer under most conditions, but it also suppresses liver fibrosis, which depends on the progress of liver injury and the cell types in which the STAT3 are activated.
3The role of STAT3 in NAFLDNAFLD is becoming the most prevalent liver disease in the western world, with a prevalence of 20%.
In a subgroup of patients, the pathological accumulation of fat in the form of triglycerides in hepatocyte lipid droplets is a histological hallmark of NAFLD, such as ballooning degeneration of hepatocytes with inflammation, and also by a varying degree of fibrosis. Histologic features are advanced liver fibrosis (stage F3) and cirrhosis (stage F4) [28,29].
Accumulated research data reveal that, whereas blockading of hepatic STAT3 activation via other substances prevented NAFLD-induced liver fibrosis, STAT3 activation is also a survival signal that protects against lipotoxicity.
STAT3: an anti- and pro-inflammatory signal in NAFLD
3.1Pro-inflammatory signalAccording to recent studies, liraglutide is currently considered an effective treatment for type 2 diabetes mellitus and is expected to diminish the fatty degeneration and fibrosis of the liver. The investigators found that liraglutide treatment modulated primary Kupffer cells to M2-like activation via the cyclic adenosine monophosphate -protein kinase A (PKA)-STAT3 signaling pathway in wild mouse livers. The PKA inhibitor, H-89, was responsible for exploring the signaling pathways related to macrophage polarization. The results revealed that the expression of p-STAT3 returns to lower levels in palmitic acid-stimulated Kupffer cells after the treatment of liraglutide and the PKA inhibitor H-89 [30]. It has been clearly documented that a combination of blueberries and probiotics (BP) can effectively dampen liver inflammation [31,32]. Zhu et al. revealed that BP attenuates NAFLD, partly by inhibiting the apoptosis of hepatocytes via reducing the IL-22-mediated JAK1/STAT3/BAX signaling pathway, which dampens the occurrence of liver fibrosis [31]. Caffeol is a diterpene characteristic of coffee. It is found in coffee beans and has anti-angiogenic and anti-inflammatory effects, while it inhibits liver fibrosis. Investigator demonstrates that kahweol represses the lipopolysaccharide (LPS)-induced production of IL-1α, IL-1β, IL-6, and tumor necrosis factor-α in primary KC, primary HC, and their co-cultures. Interestingly, they found that the inhibitory effect of kahweol on liver inflammation is related to the diminishment of the expression of LPS-stimulated phospho-nuclear factor kappa beta (NF-κB) and phospho-STAT3 [33]. To evaluate the clinical function of C188-9, which is a small molecule inhibitor of STAT3, Jung et al used a NASH-associated HCC model. Analysis of gene expression indicated that the downstream signaling pathways of STAT3, STAT1, Triggering Receptor Expresses on Myeloid Cells-1, and Toll-like receptors were inhibited by the treatment of HepPten-mice with C188-9, resulting in resistance to the pathological lesions of NASH, which consequently alleviated the liver fibrosis [34].
3.2Anti-inflammatory signalAccumulating evidence also confirms that STAT3 in NAFLD acts as a critical anti-inflammatory signal to control NAFLD-induced liver inflammation and fibrosis. According to recent reports, these data provide a rationale for target STAT3 for the improvement of fatty liver.
Stanniocalcins (STC1 and STC2) are calcium/phosphate-regulating hormones generated by the corpuscles of Stannius [35]. Zhao et al reported that STC2 was observed in an important metabolic regulator in the liver, which resulted in the improvement of hepatosteatosis and hypertriglyceridemia in the livers of leptin-deficient and high-fat diet-induced obese mice through activation of the STAT3 signaling pathway.
Several cytokines are known to regulate hepatic triglyceride homeostasis by activating the STAT3 pathway, which inhibits liver fibrosis [35]. For instance, treatment with IL-6 ameliorated fatty liver by inducing phosphorylation of STAT3 in ob/ob and high-fat diet-induced obese mice [36], whereas the absence of IL-6 or hepatic STAT3 resulted in steatosis and hepatocellular injury in IL-10 knockout mice [37]. This confirms that STAT3 has an anti-injury effect in the liver. In addition, IL-22 also can ameliorate NAFLD through enhanced STAT3 activation in hepatocytes [38,39]. Several studies also document that both the MAPK and JAK/STAT3 signal transduction pathways are required for the activation of IL-6 in NAFLD [40]. Furthermore, the overexpression of p-STAT3 activated by IL-6 reversed palmitate-induced lipotoxicity through increasing autophagy7 and decreasing endoplasmic reticulum stress, which controls NAFLD-induced liver fibrosis [41]. It has been observed that STAT3 resists hepatocellular damage, which is characterized as a cell survival signal in many rodent models of liver injury [42–44].
Interestingly, betulinic acid or betulin might exhibit a therapeutic effect on acute ethanol-induced fatty liver via decreasing the expression of Toll-like receptor 4 and increasing the phosphorylation of STAT3 in mice and in HSCs, which is expected to be an ethanol-induced fatty liver treatment [42]. In addition to betulinic acid or betulin, puerarin also has an antifibrotic effect in NAFLD. Puerarin can attenuate NAFLD by improving leptin signal transduction via facilitating JAK2/STAT3 pathways [45]. These data serve as a rationale to select the STAT3 as the target for the treatment of NASH. However, researchers should be cognizant of the potential for unrestrained STAT3 activation to promote inflammation and cancer [46,47].
4The role of STAT3 in schistosomiasisSchistosomiasis is a parasitic disease of major public health significance that seriously harms human health. Three major species of schistosomes infect humans worldwide: Schistosoma (S.) mansoni, S. haematobium, and S. japonicum. The disease affects 200 million people worldwide and causes severe clinical symptoms [48]. The most closely relevant in liver fibrosis and HCC are S. mansoni and S. japonicum. The main pathological lesions of S. japonicum are the formation of egg granulomas in the liver and intestine, which can lead to severe liver fibrosis [49,50].
STAT3: an anti- and pro-inflammatory signal in schistosomiasis
4.1Pro-inflammatory signalSTAT3 and c-Jun are proto-oncogenes or transcription factors whose activation facilitates the initiation and progression of dysplasia during HCC tumorigenesis [51,52]. Roderfeld et al indicated that STAT3 and c-Jun, as well as DNA repair, were activated by an extract from soluble schistosome egg antigens (SEA) and culture supernatants of live schistosome egg in hamster model and cell culture experiments. Thus, STAT3 induces the occurrence of liver fibrosis and even HCC [51].
MDSCs are a heterogeneous population of immature myeloid cells that promote tumor progression by inhibiting the function of T cells, especially CD8 + T cells. Recent research found that STAT3 accelerated MDSC proliferation via promoting the expression of the calcium-binding pro-inflammatory proteins-S100A8. Yang et al demonstrated that SEA and soluble adult worm antigen could enhance the accumulation of MDSCs in S. japonicum–infected mouse spleen by inducing the reactive oxygen species production via JAK/STAT3 signaling, which accelerates tumor progression [53]. MicroRNAs are a type of endogenous, small noncoding RNA molecules that regulate the translation and transcription of many genes [54–56]. Yang et al suggested that although in the course of schistosome infection, both miR-146a and miR-146b were upregulated, only miR-146b could result in a series of Th2 cytokines related to the chronic stage of infection via facilitating STAT3/6, which induces the occurrence of liver fibrosis [54]. Therefore, the expression of STAT3 accelerates liver injury and fibrosis.
4.2Anti-inflammatory signalInterestingly, according to recent reports, additional studies have found that STAT3 could also play key roles in controlling liver injury. Recent research has promoted the idea that the induction of aging in activated HSCs might dampen hepatic fibrosis [57,58]. SEA is a complex mixture composed of a number of egg antigens. Sjp40 (S. japonicum egg antigen p40) could significantly improve hepatic stellate LX-2 cell activation and trigger cell caducity. Moreover, Sjp40-induced caducity was regulated by the STAT3/protein 53/protein 21 signaling pathway in LX-2 cells [57]. Thus, cellular senescence modulated by STAT3 could also play a vital suppressive role in liver fibrosis.
A growing body of evidence suggests that activation of STAT3 has been detected in many liver fibrosis caused by schistosome infection. However, the anti-or pro-inflammatory molecular mechanisms that explain the roles of STAT3 in the initiation and progression of fibrosis are still poorly understood, which is dependent on the cell types in which the STAT3 are activated, and the process of disease progression. The detailed effects and mechanisms of STAT3 need further research.
5The role of STAT3 in chemical liver injuryThe liver is the original position of contact for many kinds of orally-ingested therapeutic drugs, alcohol, and other xenobiotics from intestinal absorption, thus making it particularly vulnerable to chemical-induced damage. The range of chemical-induced liver diseases is wide, including dose-dependent hepatotoxicity, other cytopathic toxicities, and acute steatosis. Other known types of chemical-induced liver injury have been detected in many models. These include acute and chronic hepatitis, granulomatous hepatitis, cholestasis with bile duct injury, cholestasis with or without hepatitis, steatohepatitis, vascular disorders, and tumors. The pathological degree of chemical resulting in liver injury varies from tiny nonspecific changes in hepatic structure and function to acute liver injury, fibrosis, and liver cancer [59].
STAT3: an anti- and pro-inflammatory signal in chemical liver injury
5.1Pro-inflammatory signalCarbon tetrachloride 4 (CCl4), a hepatotoxin, is the most frequently used liver carcinogen in models to simulate human liver fibrosis and lesions in mice and rats [60]. Several known STAT3 inhibitors, designed to directly target hyper-phosphorylated STAT3 in cells, have been used as chemical probes to certify the role of STAT3 in CCl4-induced liver fibrosis.
For example, STX-0119 is the inhibitor STAT3 dimerization. Choi et al suggested that STX-0119 plays a key role in controlling the development of CCl4 and thioacetamide-induced liver fibrosis via diminishing the activation of HSCs [61]. In addition, HJC0123, a novel STAT3 inhibitor, was discovered using a fragment-based drug design method. It is considered as a survival signal that resists liver fibrosis, which inhibited the phosphorylation, nuclear translocation, and transcriptional activity of STAT3 [62]. Furthermore, rottlerin, which was originally identified as an inhibitor of protein kinase C delta could inhibit CCl4-induced fibrosis by reversing the increased levels of phosphorylated JAK2 as well as the levels of total and phosphorylated STAT3 [63].
A majority of Chinese herbal medicines that show inhibitory activity toward STAT3 activation also reduced markers of CCl4-induced liver fibrosis, indicating its effective target for treatment in liver fibrosis. CCM111 is extracted from Antrodia cinnamomea, which has a protective liver fibrosis function. Lin revealed that CCM111 restores the activity of transforming growth factor-β (TGF-β) signaling pathways to the lower level through inhibiting the levels of phosphorylated mothers against decapentaplegic homolog (Smad)2 and Smad3 and decreasing the expression of α-smooth muscle actin, matrix metalloproteinases 2, and TGF-β1. CCM111 also reduced the activity of STAT3 and Wnt pathways via inhibiting the expression of phosphorylated STAT3 and the translocation of β-catenin in HSCs-T6. CCM111 inhibits CCl4-induced expression of ECM proteins by diminishing the activation of the TGF-β, Wnt, and STAT3 pathways and by reducing inflammatory responses [64]. Asiatic acid (AA), a bioactive compound extracted from the Centella asiatica, has multiple hepatoprotective pharmacologic effects, such as antioxidative and anti-inflammatory activities [65–67]. Fan et al. discovered that AA ameliorates CCl4-induced liver fibrosis in rats by regulating nuclear factor erythroid-2 related factor 2 (Nrf2)/antioxidant response element, NF-κB/inhibitor of NF-κB α, and JAK1/STAT3 signaling pathways, indicating it might be a new therapeutic approach aimed at liver fibrosis [65].
S-allyl-cysteine (SAC) is an extract from major compounds in aged garlic, which plays protective roles in the gastrointestinal tract and liver. SAC attenuated CCl4-induced liver fibrosis in rats with anti-oxidant, anti-inflammatory, and anti-fibrotic effects. Furthermore, SAC reduced the phosphorylation of SMAD3 and STAT3, and further inhibited their ability of binding to transcription promoters [68], which suggests that SAC might be a new antifibrosis target that improves liver fibrosis. In addition to SMAD3, other evidence suggests that STAT3 contributes directly to the activation of HSCs and transdifferentiation in response to TGF-β and promote hepatic fibrosis [69]. Panax notoginseng saponins are characterized by their beneficial biological effects [70]. A study showed that panax notoginseng saponins can inhibit the JAK2/STAT3 signaling transduction pathway to improve CCl4-induced hepatic fibrosis in vivo and in vitro [71]. In the occurrence and development of liver fibrosis induced by CCl4, some cytokines and signaling molecules participate in the regulation of liver fibrosis. Mesenchymal stem cells (MSCs) are identified as multipotent adult stem cells by their ability to differentiate into hepatocyte-like cells [72–74]. They secrete a series of cytokines and signaling molecules that dampen inflammation, stimulate the hepatocyte proliferation, and maintain the function of liver cells [75]. Bone marrow–derived mesenchymal stem cells (BM-MSCs) might play an immunomodulatory role in the improvement of CCl4-induced liver fibrosis through the downregulation of the IL17A/IL6/STAT3 signaling pathway. However, the therapeutic effect of MSCs on different diseases is controversial. BM-MSCs might promote the IL6/STAT3 signaling pathway and facilitate cell invasion in hepatocellular carcinoma [72]. Thus, the effect of STAT3 in liver fibrosis requires further investigation. Nrf2 has been recently defined as a promising therapeutic target for hepatic fibrosis. Nrf2 facilitated the suppressor of cytokine signaling 3 expression, which inhibited JAK2/STAT3 signaling. Nrf2 was decreased in activated HSCs and negatively correlated with hepatic fibrosis severity in human liver specimens, suggesting Nrf2 might be a new target molecule that improves liver fibrosis by inhibiting the expression of STAT3 [76].
5.2Anti-inflammatory signalInterestingly, the above research has found that the induction of aging in activated HSCs might dampen hepatic fibrosis [57,58]. Huang revealed that IL-10 could induce senescence of activated HSCs via promoting the phosphorylation and translocation of STAT3 to attenuate liver fibrosis [77].
The liver is vulnerable to being damaged by chemical drugs, such as CCl4. CCl4 is one of the most common hepatotoxins inducing liver injury. Therefore, we investigated a novel potential molecular mechanism for the pathogenesis of chemical-induced liver diseases to be an effective therapy for liver fibrosis [54].
6OutlookAberrant expression of a series of acute phase proteins, chemokines, and chemokine receptors in activated STAT3 can aggravate liver inflammation in CCl4-induced liver fibrosis. However, in HBV, NAFLD, and schistosomiasis, STAT3 can act as a pro- or anti-inflammatory signal in modulating liver inflammation and liver fibrosis depending on the liver injury models being studied. Accumulated research data from the last decade have indicated that STAT3 and its related cytokines show complex biological effects in diverse factors induced fibrosis. Activation of STAT3 can play anti-or pro-inflammatory roles in the pathogenesis of liver fibrosis, dependent on the cell types in which the STAT3 are activated and the type of liver disease or liver injury model being studied. These findings have shown that the roles of STAT3 in liver inflammation and liver fibrosis are complex (Figs. 1 and 2).


In summary, an abundance of evidence demonstrates the hypothesis that STAT3 makes critical contributions to the development and progression of fibrosis in the liver. These studies also indicate that use of STAT3 inhibitors in the liver injury model shows promising anti-fibrotic activity, which highlights that STAT3 is an effective target for the treatment of fibrotic diseases in patients.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors’ contributionsJZ and YRY designed the study and revised the article. JZ reviewed the literature and wrote the article. JZ, YRY and YFQ reviewed the literature and revised the article. All authors read and approved the final manuscript.
Conflict of interestThe authors have no conflicts of interest to declare.AbbreviationsJAK Janus protein tyrosine kinase Signal transducer and activator of transcription chronic HBV Xiaochaihu decoction homeobox protein A10 enhancer element I blueberries and probiotics Stanniocalcins S-allyl-cysteine



